jstar809
Harmless

Posts: 1
Registered: 9-12-2011
Member Is Offline
Mood: No Mood
|
|
how does the ring form??
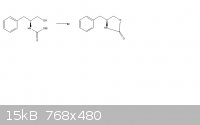
(attached is the reaction scheme)
So i'm trying to figure out how this oxazalidinone ring forms:
The reagents are Potassium carbonate, and its heated to 135 degrees celsius. My prediction is that the OEt group would leave and the alcohol would be
deprotonated by it. This will then cause a bond to form the ring structure.
Please let me know what you think, I've attached the picture.
|
|
|
Megak
Harmless
Posts: 12
Registered: 8-12-2011
Member Is Offline
Mood: No Mood
|
|
I think that the alcohol would just be liberated from the molecule and evaporate off, though my organic is weak so you should probably check with
someone else 
|
|
|
BromicAcid
International Hazard
Posts: 3227
Registered: 13-7-2003
Location: Wisconsin
Member Is Offline
Mood: Rock n' Roll
|
|
Carbamates can be pretty insensitive but 135°C can be a convincing driving force. Carbonate is good as an irreversible proton plucker although a bit
slow, possibly deprotonation of the alcohol (which doesn't have much motivation aside from the heat) followed by nucleophilic attack on the carbonyl,
tetrahedral intermediate, with carbonyl oxygen carrying negative charge and in a relationship with the K cation and finally back to the carbonyl with
splitting off of the ethoxide anion to mate with the potassium cation.
The fact that you would be going from potassium carbonate to potassium ethoxide worries me since that's against the usual order of things, right? Or
has it really been that long? So in the end you would have potassium ethoxide, potassuim hydroxide (from some of the carbonate going to H2O and CO2),
potassium bicarbonate, and ethanol (from hydrolyzed potassium ethoxide).
I don't really like the deprotonation or the attack on the carbamate, but they could happen at these temps.
|
|
|
ziqquratu
Hazard to Others
Posts: 385
Registered: 15-11-2002
Member Is Offline
Mood: No Mood
|
|
It's basically a nucleophilic acyl substitution reaction (Wikipedia has the mechanism).
The alcohol attacks the carbonyl, giving the traditional tetrahedral intermediate; proton transfer to the EtO group and subsequent loss of EtOH gives
the oxazolidinone. The reaction is driven by several factors: the close association of the nucleophile with the carbamate (due to it being
intramolecular); the formation of a ring (tends to drive many reactions, for certain ring sizes, at least); and the increase in entropy (1 mol
starting materials --> 2 mol of products (1 of oxazolidinone + 1 of EtOH)).
The reverse reaction is quite unfavourable, as it would result both in loss of entropy and ring opening, and is intermolecular (i.e. the nucleophile
is nowhere near the carbamate).
The K2CO3 could be there as a buffer, or may be serving to activate the carbamate, by association of K+ with the carbonyl oxygen (essentially like a
very mild Lewis acid). Or it may serve some other purpose I'm overlooking - but deprotonation of either alcohol (starting material or EtOH) is NOT
likely.
|
|
|
jon
Hazard to Others
Posts: 459
Registered: 11-1-2006
Member Is Offline
Mood: paranoid distrustful apprehensive
|
|
i think the carbonate pulls protons off the tetrahedral intermediate.
at least that's my theory because i use sodiuum acetate and pyridine in acylations all the time.
Give me librium or give me meth!
Patrick Henry....
|
|
|
ziqquratu
Hazard to Others
Posts: 385
Registered: 15-11-2002
Member Is Offline
Mood: No Mood
|
|
For acylations, typically using acid chlorides or anhydrides, you need a base to mop up the acid formed (e.g. HCl from acid chlorides, because the
leaving group is Cl-). Also, for those reactions, pyridine is often used to catalyse the reaction, by forming an acyl pyridinium intermediate, in
addition to its role as a base. Another common reagent for this purpose is DMAP (4-dimethylaminopyridine), and you can see the mechanism towards the
bottom of the Wiki page for the Steglich esterification.
As far as the tetrahedral intermediate goes, the proton transfers are usually intramolecular - from the incoming OH to the carbonyl oxygen, then from
there to the leaving group before or as it leaves.
I got to reconsidering, however, and I believe that it isn't so unreasonable to think that K2CO3 could, to some degree, deprotonate the alcohol -
particularly at those high temperatures. The alkoxide WOULD be a better nucleophile, and that may very well be its role. Of course, the equilibrium
would likely favour the alcohol, but you only need a little alkoxide to form, then react, then more forms (Le Chatelier's principle), and so on until
conversion is complete.
|
|
|
Arrhenius
Hazard to Others
Posts: 282
Registered: 17-8-2008
Location: US & A
Member Is Offline
Mood: Stochastic
|
|
Hi all,
I thought I'd add a few things. The reaction procedure is here at Org Syn. It's run neat in diethyl carbonate.
Jstar809:
I think what you wrote is on track, but not really mechanistically insightful. See below.
BromicAcid:
| Quote: |
Carbonate is good as an irreversible proton plucker although a bit slow, possibly deprotonation of the alcohol
|
Probably not, see pKas considered below. Carbonate is a weak base, and thus reversible, as well. I think
you're aware of this, though. Also, consider that K2CO3 is insoluble in the reaction.
ziqquratu
| Quote: |
It's basically a nucleophilic acyl substitution reaction
|
A carbamate is not really an acyl group, so I'd hesitate invoking this analogy.
| Quote: |
The reaction is driven by several factors... the formation of a ring... and the increase in entropy
|
Yes, definitely. 5 and 6 member rings are kinetically favorable, as there's little or no ring strain in
the TS or product.
Anyway, here's my thoughts. The pKas are taken from the very helpful Evans pKa table.
Scheme 1: BromicAcid's mechanism. Again, the problem here is that the base is not sufficiently strong to promote this. According to
pKas, the equilibrium would lie some 10e6 in favor of the starting material. This equilibrium is not substantially temperature dependent. I do know
that in the case of N-Boc, potassium t-butoxide will promote this ring closure at room temp, and here the relative pKas will allow this. In general,
base or acid catalyzed mechanisms require the catalyst be within ~4 pKa units of the intermediate.
Scheme 2: ziqquratu's mechanism. I think this is unlikely, given that it's not possible to do a 'trans esterification' type of
reaction on carbamates (as shown, R-OH). Admittedly the high effective concentration of 5-member TS might allow this.
Scheme 3: If you consider the acidity of the carbamate NH (pKa ~12 in H2O), this deprotonation is well within the realm of a base
promoted mechanism, and we should expect some ~100:1 equilibrium favoring the protonated species. At 135º this carbamate potassium salt decomposes to
the isocyanate (entropically favorable, and known) and undergoes a 5-exo-dig cyclization (enthalpically favorable). A proton transfer and
tautomerization gets to the product.

[Edited on 11-12-2011 by Arrhenius]
|
|
|
qw098
Harmless
Posts: 22
Registered: 25-10-2011
Member Is Offline
Mood: No Mood
|
|
Hey Arrhenius, what program did you use to draw those schemes? They look awesome!
|
|
|
AndersHoveland
Hazard to Other Members, due to repeated speculation and posting of untested highly dangerous procedures!
Posts: 1986
Registered: 2-3-2011
Member Is Offline
Mood: No Mood
|
|
Looking at the picture, I would guess that the reaction proceeds through a complex series tautomerizations. Essentially, that double bond moves, one
position at a time, counterclockwise, until the original hydroxyl groups becomes an aldehyde. From there, the aldehyde can easily accept the extra
electron that would now be on the other carbonyl group.
The presence of the "toluene" group probably is important, or essential, to the reaction, as it makes it more favorable for the carbon atom (which is
also connected to the N atom) to which it is connected to transiently accept an extra electron. This has nothing to do with the benzene ring in the
"toluene", of course.
|
|
|
ziqquratu
Hazard to Others
Posts: 385
Registered: 15-11-2002
Member Is Offline
Mood: No Mood
|
|
Anders - no way that type of tautomerisation is going to occur. Those electrons are going to stay very happily somewhere between the carbamate
nitrogen and oxygen - they are not going to move along to the CHN carbon, let alone any further. And an aldehyde is DEFINITELY not going to be an
intermediate in this reaction.
As for Arrhenis' mechanism - it's not at all unreasonable. I'm not comfortable with the idea of an isocyanate-type intermediate, but it's quite
possible. Your logic regarding pKa is definitely good, however, and I would certainly agree that an intermolecular reaction of this type is
unfavourable. However...
With regards to the mechanism drawn for my proposal, one small (but important) error is that the leaving group for a nucleophillic acyl substitution
is EtOH, a far better leaving group than Bromic's EtO<sup>-</sup>...
Also, bear in mind that carbonates can most certainly be transesterified via the traditional (they cannot go through an isocyanate!) mechanism, even
using such weak nucleophiles as phenols; the same may well hold for carbamates (they're less reactive, yes, but for an intramolecular, ring-forming
reaction in a polar aprotic solvent at high temperature?).
I was going to talk my idea out, but attached a scheme instead (please ignore the fact I didn't tidy it up - quickly thrown together in ChemDraw).
Using your proposed deprotonation of the carbamate will give a imidate potassium salt (K associated primarily with the oxygen - behaving, to some
degree, like a Lewis acid), which will further activate it towards a nucleophile. Nucleophilic attack results in highly favourable proton transfer to
the nitrogen (since the N- would be a VERY strong base), followed by protonation of the alkoxide. Finally, proton transfer allows EtOH to leave,
giving the product.
I'm going to have to look into this properly when I get some time, but I stand by my original argument - nucleophilic acyl substitution, driven by a
number of factors (intramolecular, ring formation, entropy increase, and now adding Arhennius' imidate formation to activate the carbonyl).
|
|
|
AndersHoveland
Hazard to Other Members, due to repeated speculation and posting of untested highly dangerous procedures!
Posts: 1986
Registered: 2-3-2011
Member Is Offline
Mood: No Mood
|
|
I was thinking something like the below, (sorry I do not have the proper program available to me to create a picture)
CH3CH2-O-C(=O)-NH-CHR-CH2-OH
CH3CH2-O-C(-O[-])=NH-CHR-CH2-OH
CH3CH2-O-C(-O[-])=NH[+]-CHR-CH2-OH
CH3CH2-O-C(-OH)=N-CHR-CH2-OH
CH3CH2-O-C(-OH)=N-C[-]R-CH2-OH, H[+]
CH3CH2-O-C[-](-OH)N=CR-CH2-OH, H[+]
CH3CH2-O-CH(-OH)N=CR-CH2-OH
CH3CH2-O-CH(-OH)NH-CR=CH-OH
CH3CH2-O-CH(-OH)NH-CHR-CH=O
CH3CH2-O-CH(-O[-])NH-CHR-CH=O, H[+]
...-CH=O [-]O-CH(-OEt)-NH-...
...-CH(-O[-])-O-CH(-OEt)-NH-...
-CHR-CH(-OH)-O-CH(-OEt)-NH-
-CHR-CH(-OH)-O-CH(-OEt)-NH-
-C[-]R-CH(-OH)-O-CH(-OEt)-NH-
-CR=CH-O-CH(-OEt)-NH-, OH[-]
-CR=CH-O-CH(-OEt)-NH-, H[+]
-CHR-CH=O[+]-CH(-OEt)-NH-
-CHR-CH=O[+]-C[-](-OEt)-NH-
-CHR-CH[-]-O[+]=C(-OEt)-NH-
-CHR-CH2-O[+]=C(-OEt)-NH-
-CHR-CH2-O[+]=C(-OEt)-NH-, OH[-]
-CHR-CH2-O-C(-OH)(-OEt)-NH-,
-CHR-CH2-O-C(-O[-])(-OEt)-NH-
-CHR-CH2-O-C(=O)-NH-, [-]OEt
I am not saying there is not more than one possible route.
[Edited on 13-12-2011 by AndersHoveland]
|
|
|
Arrhenius
Hazard to Others
Posts: 282
Registered: 17-8-2008
Location: US & A
Member Is Offline
Mood: Stochastic
|
|
qw098
Scheme was drawn with ChemDraw 12.0 - format is ACS '96 style with line with 0.017in and bold width 0.030in. I would recommend you download ACD
ChemSketch - free student version is (or used to be) available online.
AndersHoveland
| Quote: |
I would guess that the reaction proceeds through a complex series tautomerizations.
|
Sorry, I just don't think so. There's no need to enter uncharted waters here, and I would bet one or several of the mechanisms outlined above is(are)
operative. As for accessing an aldehyde, this would almost certainly mix the stereocenter under the reaction conditions, and that's not what's
observed.
ziqquratu
| Quote: |
The presence of the "toluene" group probably is important,
|
It's not. This reaction, or a similar variant, works with other amino acid N-carbamates as well.
| Quote: |
I'm going to have to look into this properly when I get some time, but I stand by my original argument - nucleophilic acyl substitution, driven by a
number of factors (intramolecular, ring formation, entropy increase, and now adding Arhennius' imidate formation to activate the carbonyl).
|
It sounds good, but let's critically consider one problem here. The deprotonated carbamate is
less electrophilic as a result of being more electron rich! So I wouldn't recommend invoking that. I understand what you're
trying to say about protonation of the tetrahedral intermediate to allow EtOH to leave as opposed to EtO-, but I don't know what the pKa of the
tetrahedral intermediate is. Also, under basic conditions it's not really appropriate to utilize acid catlyzed steps in your mechanism. That is to
say, it's not reasonable for an intermediate to possess a pKa lower than that of the base in the reaction.
[Edited on 14-12-2011 by Arrhenius]
|
|
|
AndersHoveland
Hazard to Other Members, due to repeated speculation and posting of untested highly dangerous procedures!
Posts: 1986
Registered: 2-3-2011
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by Arrhenius  |
| Quote: |
I would guess that the reaction proceeds through a complex series tautomerizations.
|
As for accessing an aldehyde, this would almost certainly mix the stereocenter under the reaction conditions, and that's not what's observed.
|
Agreed, if the final product was not observed to be a mix of stereoisomers, with respect to the "R" group ("toluene" group), then the proposed
mechanism that I posted must be, at most, not be relevant as a significant pathway.
| Quote: Originally posted by Arrhenius |
| Quote: |
The presence of the "toluene" group probably is important,
|
It's not. This reaction, or a similar variant, works with other amino acid N-carbamates as well.
|
What was meant is that I did not think that the reaction would proceed if "R" was a hydrogen atom. But perhaps there is an example that disproves
this.
Perhaps Arrhenius is erroneous? 
[Edited on 15-12-2011 by AndersHoveland]
|
|
|
kmno4
International Hazard
Posts: 1495
Registered: 1-6-2005
Location: Silly, stupid country
Member Is Offline
Mood: No Mood
|
|
To make this refereneless discussion more strict, you may read:
Oxazolidin-2-one Ring, a Popular Framework in Synthetic Organic
Chemistry: Part 1. The Construction of the Oxazolidin-2-one Ring
| Code: | http://www.benthamscience.com/cos/sample/cos4-1/006AH.pdf |
(S)-4-(PHENYLMETHYL)-2-OXAZOLIDINONE
| Code: | http://www.orgsyn.org/orgsyn/prep.asp?prep=cv8p0528 |
|
|
|
Chordate
Hazard to Others
Posts: 108
Registered: 23-2-2011
Member Is Offline
Mood: No Mood
|
|
The first step of this reaction, the formation of the carbamate from the amine & diethyl carbonate (DEC) is most baffling to me. According to that
paper the whole reaction is accomplished at reflux (128 degrees for DEC) in 2.5 hours using no catalysts and a weak base.
All other references I can find to urethane synthesis by these means either use a very strong base, exotic catalyst, activate the carbonyl using
something like a mitsunoba reagent, or use neat K2CO3 in an autoclave at 200+C for 22+ hours. Now, granted these are only using dimethyl carbonate,
but my intuition tells me that that isn't enough of a difference to effect a 10 fold increase in reaction rate at half the temperature unless
something new to me is going on.
F'rinstance:
http://www.iupac.org/publications/pac/pdf/2005/pdf/7710x1719...
http://144.206.159.178/FT/1010/64161/1093240.pdf
http://www.sciencedirect.com/science/article/pii/S0040402011...
Once the initial addition is accomplished the rest of the reaction becomes quite easy because you are no longer relying on potassium carbonate to
deprotonate the alcohol.
Edited: screwed up my links.
[Edited on 17-12-2011 by Chordate]
[Edited on 17-12-2011 by Chordate]
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
Chordate, in the Pure Appl. Chem. article you cite, it is clearly described that methyl carbamates easily form by heating amines in dimethyl
carbonate already at 90 °C. The presence of the base accelerates this reaction, but it is not required. Mind that with diethyl carbonate the
alkylation reaction is no more feasible so that this side reaction is of no concern. Citing the authors:
| Quote: | | The behavior of amines in the presence and in the absence of a base reported on Tables 2 and 3 confirms that, since the hardness of the nucleophile is
increased while operating in the presence of a base, the BAc2 rate is dramatically accelerated, and carboxymethyl derivatives are quantitatively
obtained. |
Arrhenius, your mechanistic proposal via Scheme 3 is indeed a possibility, but since I know from experience that similar cyclizations occur also on
N-alkylcarbamate type of substrates (where isocyanates can not form) this can not be the only operating mechanism (if operating at all). The
proposal in Scheme 1 is in my opinion the most reasonable. Note that the deprotonation with K2CO3 is highly reversible and even though carbamates are
more acidic than primary alcohols, you will always have both species present in equilibrium (especially since intramolecular proton exchange in this
case is extremely rapid due to geometry).
The isocyanate intermediate is however a reality in certain related reactions. For example, in the reaction of amines with urea to give
N,N'-dialkylureas. I had to learn this by experience when trying it on secondary amines. Ammonia needs to eliminate from the N-alkylurea intermediate, which can not happen on
N,N-dialkylureas. There is also a study that demonstrates that primary amines react with COCl2 to give ureas mostly via the isocyanate intermediate
formed by HCl elimination (explaining the slower rate in the corresponding reaction with secondary amines, but it would be too much work for me to
find this reference - unless really needed).
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
Arrhenius
Hazard to Others
Posts: 282
Registered: 17-8-2008
Location: US & A
Member Is Offline
Mood: Stochastic
|
|
Heh. Nicodem beat me to it!
Chordate: I'm not really sure why you're 'baffled'. If you read your references, you shouldn't be. From Tundo et al
Pure Appl. Chem., 2005, 77(10), 1719–1725:
| Quote: |
In these conditions, high yields of carbamates are obtained in few minutes (Table 3). Since aliphatic amines are harder nucleophiles than aromatic
ones, they react faster with the carbonyl of DMC (entries 3–7). No N,N-dimethyl derivates were observed.
|
Phenethylamine reacts with dimethylcarbamate at 90ºC in the presence of methoxide to give the
methylcarbamate in 100% yield in 5 min.
| Quote: |
Weaker bases like potassium carbonate can be used instead of alkoxides... In these conditions, harder amines react in 22 h and give the corresponding
carbamates and
N-methylcarbamates, while mono- and dimethyl derivatives, if any, are present in lower amount.
|
They're only using an autoclave because the boiling point of dimethylcarbonate is lower. Again,
phenethylamine gives a 77% yield under these conditions.
Nicodem
Agreed, there is always the possibility for multiple reaction pathways, and it becomes a relative rate issue. Good point on N-alkyl carbamate ring
closure. My only thought here was that by N-alkylating you may favorably bias the amide rotamer. In all of my drawings, the amide is s-trans - easily
accessed at 127ºC - but if the substrate is dialkyl, and the nucleophile is favored s-trans maybe this would accelerate direct addition-elimination.
|
|
|
Chordate
Hazard to Others
Posts: 108
Registered: 23-2-2011
Member Is Offline
Mood: No Mood
|
|
Wait wait, I'm not saying it is impossible! It is clearly happening, I am just wondering why that the rate is so accelerated. Like you said, the
reaction proceeds with 100 percent yield in the presence of methoxide in five minutes, but takes TWENTY TWO HOURS with dimethyl carbonate and the
weaker base K2CO3?
Why is the .4 pKa difference between the methoxide leaving group and the ethoxide leaving group making the reaction around 15 times faster? Is it just
because they are distilling off the ethanol or what?
[Edited on 18-12-2011 by Chordate]
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
It takes 2.5 h at bellow 126 °C to achieve both, the carbamate formation and ring closing, in the Org. Synth. example. It is not clear if
the carbamate formation is faster than ring closure, but in any case I would say 2.5 h with 10 mol% K2CO3 (heterogeneous conditions) are comparable to
3-10 min with 1.2 equivalents of NaOMe at 90 °C with dimethyl carbonate as reported by Tundo et al. I would say that they heated for 22 h in their
examples with K2CO3 because seeing that the reaction is not selective enough for the carbamate, they went to further conversion to see what mixture
they get. Keep in mind that with dimethyl carbonate you have also the N-methylation reactions to observe and record.
|
|
|
kmno4
International Hazard
Posts: 1495
Registered: 1-6-2005
Location: Silly, stupid country
Member Is Offline
Mood: No Mood
|
|
OS conditions are homogeneous:
...preheated to 135°C, and is stirred until dissolution is achieved (ca. 5 min)
Reference [39] from Bentham paper is worth reading.
Here is picture extracted from this paper:
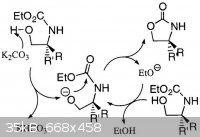
BTW.
Example of transesterification (no carbamates): Activated Carbon Supported K2CO3 Catalysts for Transesterification of Dimethyl Carbonate with
Propyl Alcohol, DOI: 10.1021/ef060492m
[Edited on 18-12-2011 by kmno4]
|
|
|












