| Pages:
1
..
8
9
10
11
12
..
19 |
Formatik
National Hazard

Posts: 927
Registered: 25-3-2008
Member Is Offline
Mood: equilibrium
|
|
It's commercial. I haven't oxidized SO2 or NO2. Oxidizing SO2 with H2O2 is less hassle and simpler and that I have done, but had some problems with
the SO2 generator, so quit the experiment (H2SO4 thread). With SO2 at least I expect the reaction to be quite exothermic. Dry O3 will not react with
SO2, but in the presence of H2O immediate oxidation to H2SO4 occurs according to Schönbein (J. pr. Ch. 95 [1865] 469/79, 472; etc). Liquid O3 doesn't
react with liquid SO2, which might be hard to believe but that was established by Briner, Lachmann (Helv. chim. Acta 26 [1943] 346/57).
The reaction of O3 with nitrogen oxides can be more complex. Some reactions with NO, NO2, N2O5: Leading NO into liquid air cooled liq. O3-O2 mixture
will form a green compound (same one from leading NO into liquid air), which explodes as the solution evaporates. Blowing N2O4 into O3-containing O2
forms N2O5, gives also a blue coloring. Rapid leading in of O2-O3 mixture at 0 deg. over the surface of liq. N2O4 gives a blue flame at the end of the
inlet tube which sometimes travels through the weakly blue gas leaving behind brown NO2 - Lemon, Lowry (J. chem. soc. 1936 1409/12). N2O5 and O3 give
NO3 containing bluish gas at cool temps (0 deg.) which can be ignited.
It might just be my view on it, but frankly, I think ozone is worthless for synthesis. The operations take too long or are low efficiency, or plagued
by other complications. Example: bubbling ozonated air into 75 mL 10% NH4OH in a slightly covered (to allow some recirculation) 1L RB flask for 2.7
hours. Although clearly some white NH4NO3 fog was visible after the operation, the solid recoverable was: 0.05 to 0.1g. Not counting loss of NH3 by
evaporation. Maybe the water interfered a lot here, since ozone is barely soluble it probably required the O3-air current to push NH3 out of the
solution in order to react with it (in the gas phase).
[Edited on 28-4-2009 by Formatik]
|
|
|
Alexein
Harmless
Posts: 35
Registered: 14-7-2007
Member Is Offline
Mood: Metastable
|
|
Copper based nitric acid prep
3 part video on "home" nitric acid synthesis.... home if you happen to have sophisticated glassware... ah well.
http://www.youtube.com/watch?v=2yE7v4wkuZU
The first 2 methods are variations on a copper based approach. Horribly expensive (in terms of cost/yield ratio) but a simple approach nonetheless.
The last method is the classic sulfuric acid & nitrate salt distillation prep.
|
|
|
User
Hazard to Others
Posts: 339
Registered: 7-11-2008
Location: Earth
Member Is Offline
Mood: Passionate
|
|
Ah well so expensive does it not have to be.
I mean there is so much scratch copper laying around.
And if you have your sources HCl is almost for free.
I like the idea.
If it is so very practical , thats a second thing.
What a fine day for chemistry this is.
|
|
|
497
National Hazard
Posts: 778
Registered: 6-10-2007
Member Is Offline
Mood: HSbF6
|
|
It seems to me that the reaction between a persulfate salt and N2O4 could yield fairly pure and concentrated HNO3 via:
N2O4 + H2O --> HNO3 + HNO2
then
Na2S2O8 + H2O + HNO2 --> 2NaHSO4 + HNO3
overall:
Na2S2O8 + 2H2O + N2O4 --> 2NaHSO4 + 2HNO3
Why wouldn't this work? Persulfate should be a more than strong enough oxidizer to oxidize NO2- to NO3-.
It seems to me it would be possible to add just a near stoiciometric amount of water to yield very concentrated HNO3. Unless it wouldn't run to
completion without excess H2O for some reason? I don't know.
EDIT;
Another related possibility is potassium peroxymonosulfate (aka Oxone) which is available quite cheaply as a pool chemical. Maybe like so:
KHSO5 + H2O + N2O4 --> KHSO4 + 2HNO3
[Edited on 4-6-2009 by 497]
|
|
|
DJF90
International Hazard
Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
Persulfate is a kinetically hindered oxidant; it is very strong but also quite slow. A catalytic amount of silver (I) can be used to greatly improve
its oxidative speed by froming AgO as the oxidising species. This should also not affect the production of nitric acid in the way that you suggest.
[Edited on 4-6-2009 by DJF90]
|
|
|
lopos123
Harmless
Posts: 14
Registered: 25-3-2009
Member Is Offline
Mood: No Mood
|
|
I don't know if this sounds interesting, but i've found this in a chemistrybook.
Picture
[Edited on 4-6-2009 by mikkello]
|
|
|
User
Hazard to Others
Posts: 339
Registered: 7-11-2008
Location: Earth
Member Is Offline
Mood: Passionate
|
|
Sure does, if the amounts produced are of any use at all.
And if there are no heavy contaminations.
Bisulfates and NO3 salts are cheap ass.
I've just tried it and indeed it releases densy brown fumes.
The big issue would be that when it is heated decomposition directly effects the output.
[Edited on 4-6-2009 by User]
What a fine day for chemistry this is.
|
|
|
cobrasniper555
Harmless
Posts: 20
Registered: 6-9-2007
Location: Tucson, AZ
Member Is Offline
Mood: A pretty good mood!
|
|
@Formatik: Very cool! I have been doing research on laboratory run at the Ostwald process for a while now and have thought about performing this
experiment sometime soon. I am very glad to see that both you and kilowatt have pictures and/or video on the subject. However, I do have a question.
Would it be possible to bubble the NO2 through a basic solution of potassium bicarbonate or something similar? I don't want to worry about the
concentration of the HNO3 dissolved, but rather have some KNO3 form within the reaction vessel. If the concentration of the base is low enough within
the water, and the water is cool, then wouldn't the KNO3 be precipitated out, instead of dissolving as well? This would surely make collecting easier.
If pro is the opposite of con, shouldn\'t Congress be the opposite of progress?
-------------------------------------------
\"The government solution to a problem is usually as bad as the problem.\" - Milton Friedman
|
|
|
hissingnoise
International Hazard
Posts: 3940
Registered: 26-12-2002
Member Is Offline
Mood: Pulverulescent!
|
|
Bubbling NO2 into a basic solution will give a mixture of nitrite and nitrate. . .
|
|
|
User
Hazard to Others
Posts: 339
Registered: 7-11-2008
Location: Earth
Member Is Offline
Mood: Passionate
|
|
I am not sure about this but might adding a strong oxidizer prevent this?
Besides i can imagine making your own KNO3 from almost thin air is cool, but in what way would this be useful ?
Why are there so little people experimenting with the ostwald process, I mean even if it would produce low yields it would be great.
[Edited on 13-6-2009 by User]
What a fine day for chemistry this is.
|
|
|
hissingnoise
International Hazard
Posts: 3940
Registered: 26-12-2002
Member Is Offline
Mood: Pulverulescent!
|
|
Well, because the catalysts are ridiculously expensive for one thing. . .
And because getting purified NH3 to the catalyst at the correct temperature together with a stoichiometric volume of O2 is just too difficult.
The absorption towers represent the final straw. . .
|
|
|
User
Hazard to Others
Posts: 339
Registered: 7-11-2008
Location: Earth
Member Is Offline
Mood: Passionate
|
|
I just like to be open minded, maybe iam a dreamer.
A pt screen or wire is an investment that if used with a little care lasts a lifetime.
Well even if both reagents are way of stoichiometric amounts I can imagine the process still runs, or is it so fragile ?
What a fine day for chemistry this is.
|
|
|
DJF90
International Hazard
Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
With excess oxygen I can see no reason why nitric would not be produced. With excess ammonia, ammonium nitrate would form which may produce problems
(clogging of orifices etc), beside meaning nitric acid would not be the product.
|
|
|
Formatik
National Hazard
Posts: 927
Registered: 25-3-2008
Member Is Offline
Mood: equilibrium
|
|
Quote: Originally posted by cobrasniper555  | | @Formatik: Very cool! I have been doing research on laboratory run at the Ostwald process for a while now and have thought about performing this
experiment sometime soon. I am very glad to see that both you and kilowatt have pictures and/or video on the subject. However, I do have a question.
Would it be possible to bubble the NO2 through a basic solution of potassium bicarbonate or something similar? I don't want to worry about the
concentration of the HNO3 dissolved, but rather have some KNO3 form within the reaction vessel. If the concentration of the base is low enough within
the water, and the water is cool, then wouldn't the KNO3 be precipitated out, instead of dissolving as well? This would surely make collecting easier.
|
Well, you will also have some NO in there. This doesn't react with either aq. NH3 or bicarbonate in solution. Definetly not fast enough if it does.
It should react with a stronger base like aq. NaOH. Bubbling NO2/NO into the aq. hydroxide will give you NaNO3 and NaNO2, with NaNO3 predominating.
| Quote: Originally posted by User | | Why are there so little people experimenting with the ostwald process, I mean even if it would produce low yields it would be great.
|
The Ostwald process uses high pressure, that's tricky to pull off in a laboratory setting. This is also just one reason why lab preparations you read
about in the books use a nitrate salt and sulfuric acid. You're supposed to also cool the NO/air or oxygen mixture so that NO2 conversion is favored.
Though the Ostwald process actually even today is the prime industrial method of nitric acid production.
|
|
|
cobrasniper555
Harmless
Posts: 20
Registered: 6-9-2007
Location: Tucson, AZ
Member Is Offline
Mood: A pretty good mood!
|
|
I thought about that right after I posted, I was just thinking of materials and chemicals I already have. After a lengthy search on the Ostwald
process, I've found this video link: http://www.youtube.com/watch?v=OcPchpqer04&feature=relat... . It is very interesting because the instructor seems to pump oxygen in there as
well. I'm thinking the same setup with an oxygen generator (2H2O2 -MnO2-> O2 + 2H2O) pumping in oxygen.
The picture below builds off of the video link I provided. As for the solution that the NO2 (considering that the reaction vessel works
properly)bubbles through, I am thinking some distilled water or a 3% H2O2 solution. I’m sure there’s many flaws in this design, because I’m
looking for a cheap way to do it. I have a very expensive and well-equipped set but this is just how I prefer things. So please, provide any criticism
that you have. 
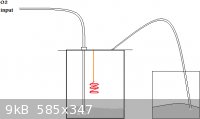
If pro is the opposite of con, shouldn\'t Congress be the opposite of progress?
-------------------------------------------
\"The government solution to a problem is usually as bad as the problem.\" - Milton Friedman
|
|
|
User
Hazard to Others
Posts: 339
Registered: 7-11-2008
Location: Earth
Member Is Offline
Mood: Passionate
|
|
I like the simplicity, it proofs that even a very primitive setup does work (neglecting the extend).
Well the receiver has to be a hell of a lot more efficient then this.
Are you planning on making a continues process?
What a fine day for chemistry this is.
|
|
|
cobrasniper555
Harmless
Posts: 20
Registered: 6-9-2007
Location: Tucson, AZ
Member Is Offline
Mood: A pretty good mood!
|
|
Not as this setup. I want to make a continuous process later on, but this is more of a temporary stage. Think of it as fundamentals towards a new
project. I'll develop new plans eventually, if this attempt at a "proof of concept" succeeds. The receiver is very simply drawn but I'll fortify it to
keep pressure and maybe recycle the gases. Then again, these would just be additions and improvements upon completion and success of this simple
setup. I'll try it later today to see how well it works. I suppose I'll post results if any are interested.
If pro is the opposite of con, shouldn\'t Congress be the opposite of progress?
-------------------------------------------
\"The government solution to a problem is usually as bad as the problem.\" - Milton Friedman
|
|
|
Formatik
National Hazard
Posts: 927
Registered: 25-3-2008
Member Is Offline
Mood: equilibrium
|
|
| Quote: Originally posted by cobrasniper555 | | I’m sure there’s many flaws in this design, because I’m looking for a cheap way to do it. I have a very expensive and well-equipped set but
this is just how I prefer things. So please, provide any criticism that you have. |
Alright. The process isn't very efficient, because some of the moist unreacted NH3 will mix with react with the formed NO2. Converting NO to NO2 is
also frustrating under regular working conditions (the reaction is also slow). The catalyst contact time of the reaction gases in the actual process
is also very short to reduce undesirable side reactions (USP3927182 explains the basics, that's a good reference to look at).
[Edited on 14-6-2009 by Formatik]
|
|
|
grndpndr
National Hazard
Posts: 508
Registered: 9-7-2006
Member Is Offline
Mood: No Mood
|
|
Speaking of PRIMITIVE setups I just lost an e-bay NOS corning brand pyrex retort marked with model # capacity 250ml and maker Inside original
packaging at less cost than modern chinese junk.Bid 25 lost it at $29had high bid plus a cushion must have ganged bids last hour.$43 w shipping for a
high quality piece versus the same size chin junk for $51+shipping.
right size for the small amounts of nitric I could have used.
An excuse to move up to real equipment,24/40 jointed columns,collectors.3way dist adapters therm. etc.
[Edited on 15-6-2009 by grndpndr]
|
|
|
DJF90
International Hazard
Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
Quickfit is much more versatile than a retort, and you'll probably feel alot better about the whole issue when you get a nice set  Unfortunately it is expensive, and takes a while to build up a decent quantity. Unfortunately it is expensive, and takes a while to build up a decent quantity.
|
|
|
entropy51
Gone, but not forgotten
Posts: 1612
Registered: 30-5-2009
Member Is Offline
Mood: Fissile
|
|
grndpndr, sorry you lost the retort on e-bay. There's something very alchemic about making HNO3 the same way it was made 3 centuries ago.
I have plenty of standard taper glass, but for making HNO3 or Br2 I dust off one of my retorts. It's a shame that Pyrex retorts are becoming so
difficult to find.
|
|
|
cobrasniper555
Harmless
Posts: 20
Registered: 6-9-2007
Location: Tucson, AZ
Member Is Offline
Mood: A pretty good mood!
|
|
@Formatik: May I ask, how is your setup going? I've developed a different method very similar to yours but with a "return" assembly to return the NO
gases, when NO2 is dissolved in water, to the original reaction vessel. I also plan on maybe increasing the surface area of the copper in the main
reaction vessel. I'm really big on this subject because if it becomes efficient, then this process may replace the constant purchasing of stump
remover.
Thanks.
If pro is the opposite of con, shouldn\'t Congress be the opposite of progress?
-------------------------------------------
\"The government solution to a problem is usually as bad as the problem.\" - Milton Friedman
|
|
|
Formatik
National Hazard
Posts: 927
Registered: 25-3-2008
Member Is Offline
Mood: equilibrium
|
|
I haven't worked on it since, it was really just a probe to see if ammonia oxidation is even successful under such conditions. To maximally optimise,
one would have to most closely approach what is possible for conversion conditions, as the Ostwald process, in the industry at least is highly
efficient.
|
|
|
froot
Hazard to Others
Posts: 347
Registered: 23-10-2003
Location: South Africa
Member Is Offline
Mood: refluxed
|
|
Just reading through the Perchloric acid topic Philou Zrealone mentions a reaction that may be worth considering for the production of concentrated HNO3 - or perhaps more
correctly concentrating dilute HNO3 in a round about way.
| Quote: | | H-C#C-H + 2AgNO3 --> Ag2C2 + 2HNO3 |
AgNO3 can be wetted with 1 or 2 drops of water and then placed in an atmosphere of C2H2 to initiate the reaction, afterwhich C2H2 can be bubbled
through until the reaction is complete. AgNO3 can be synthesised in dilute HNO3, the only trick would be to extract the Ag back out of the Ag2C2 in a
controlled manner. Working with Ag2C2 in small batches while it's wet should be much less risky. The only question would be weather the
acetylene/AgNO3 reaction would go ahead in extremely acidic conditions.
Any thoughts?
[Edited on 26-6-2009 by froot]
We salute the improvement of the human genome by honoring those who remove themselves from it.
Of necessity, this honor is generally bestowed posthumously. - www.darwinawards.com |
|
|
CMOS
Harmless
Posts: 14
Registered: 27-2-2009
Member Is Offline
Mood: No Mood
|
|
Can I use NH4NO3 to making HNO3 ?
|
|
|
| Pages:
1
..
8
9
10
11
12
..
19 |









