| Pages:
1
..
5
6
7
8
9
10 |
Hennig Brand
International Hazard

Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
I think I have a fairly safe handling process established for hydrogen sulfide gas, but I did poison myself a little during the learning process. The
danger would of course be much greater for someone not familiar with the processes and the nature of the toxin.
Your method does sound interesting and safe. Have you tried it? How sure are you that it will work?
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
The use of ferrous sulfate is reported but using a different method. The use of the sodium picrate and sodium hydroxide as the neutralizer of the
sulfate is probably best and would permit the reduction to be run at higher temperature since there is no volatility loss like could be there for the
ammonium. So I am inclining more towards use of the sodium compounds as I think about it. And I don't think the sodium sulfate byproduct would be
interfering. The reported method using ferrous sulfate was using a barium hydroxide as the base which would sequester the sulfate precipitated as
insoluble barium sulfate, but I don't think that is necessary. I think it would work for sure, it is simply a matter of working out the
concentrations for easiest manipulation of the process. Working with moderately heated and near saturation concentrated solutions I think is probably
the way to go even if there is a suspension of some undissolved material during the reduction phase, and then it can be diluted some to facilitate
filtering and isolation. Reductions seem to go better in concentrated and warm conditions generally and I expect this reduction would be no different.
The extremely low solubility of the free picramic acid should make isolation of the end product easy.
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Quote: Originally posted by Rosco Bodine  | Ferrous sulfate is cheap and safe.
9 moles FeSO4 solution and 18 moles NH4HCO3 or NH4OH or perhaps just use NaOH solution added separately and simultaneously in a parallel proportional
molar addition with stirring per mole of warm 30C sodium picrate solution should be about right for the reduction, with subsequent raising to maybe
80C and decanting and filtering of the hot solution, and acidifying to slight acidity and near complete decolorizing of the solution to precipitate
the only slightly soluble free picramic acid. The picramates are color dyes so the endpoint is visual on the acidification. I better check my
stoichiometry because I could be off and just did this one in my head. Don't bother checking the literature because it probably isn't there. 
The strategy that would be best I think is to calculate a drip rate for the ferrous sulfate into the slightly basic sodium picrate solution and then
make the additions of ammonium bicarbonate as solid sprinkled into the stirred mixture at a rate that stays slightly ahead of the ferrous sulfate to
keep the reaction mixture slightly basic. Similarly if using a liquid base the additions of base should lead by a small amount the ferrous sulfate
addition in order to keep the reaction system alkaline. Whether magnetic stirring may be good here or not is unknown since the byproduct Fe3O4 is
going to be magnetic. The magnetic interaction could be helpful or interfering, but my best guess it could be helpful and may present no issue.
[Edited on 25-3-2014 by Rosco Bodine] |
I should amend my earlier "first guess" stoichiometric ratios on this to 6 moles of ferrous sulfate being needed for reduction since the reported
byproduct of the reduction is the red iron oxide Fe2O3 instead of the mixed ferrous-ferric Fe3O4 byproduct I was guessing would be the byproduct. It
seems possible that some of both could form as the byproduct and if this is true then the actual amount of ferrous sulfate required could be a bit
more than the 6 moles but less than the 9 which would be the worst case requirement.
Reading the period literature from 150 years ago for clarification on the terminology is helpful.
The reference by Aime Girard posted earlier here
http://www.sciencemadness.org/talk/viewthread.php?tid=433&am...

Terms that are archaic "protosulphate of iron" is ferrous sulfate and the "protoxide" is ferrous hydroxide and the abundant precipitate of "peroxide
of iron" is Fe2O3 or red iron oxide which would likely form first as a perhaps gelatinous transiently stable hydrated ferric oxide then decomposing in
the hot reaction mixture to the neat oxide which should be a particulate precipitate. Girard's mention of the "ammoniacal liquid" would indicate
that the base being used was ammonium hydroxide or ammonium bicarbonate. But it would seem that other bases could be used. Sodium hydroxide should
serve as well. Milk of lime has been described in the literature and has the barium hydroxide also been used for precipitation of the sulfate value
from the ferrous sulfate. There could be advantage to removal of the sulfate value from conversion of the ferrous sulfate to ferrous chloride, using
calcium chloride and filtering out the byproduct calcium sulfate. The needed amount of hydrated lime could be neutralized with HCl and then the
solution of CaCl2 and FeSO4 could be mixed and the FeCl2 solution from filtration could be used for reduction, gradually added to a hot solution of
sodium picrate, with an added 2 equivalents of sodium hydroxide, with the byproducts of reduction being sodium chloride remaining in solution, and a
mixed precipitate of ferric oxide and sodium picramate. In the alternative using NH4OH as the base could have advantage due to the much greater
solubility of the NH4Cl byproduct than NaCl. Additional dilution water could be added and the completed reaction mixture heated to dissolve the sodium
picramate, decanted and filtered. Addition of HCl should precipitate the nearly insoluble free picramic acid from the residual NaCl or NH4Cl
solution.



The reaction appears to be
C6H2(NO2)3OH + NaOH ----> C6H2(NO2)3ONa + H2O
( 6 X ) [ FeSO4 + 2 NaOH ----> Na2SO4 + Fe(OH)2 ]
6 Fe(OH)2 + C6H2(NO2)3ONa ----> C6H2(NO2)2NH2ONa + 5 H2O + 3 Fe2O3
To provide an excess of theory of the reducing agent to compensate for atmospheric oxidation and other possible process depletion losses it would
probably be fine to use a 7 X quantity of the reactants for the second equation and use a couple of per cent excess of theory beyond for the NaOH to
assure the reaction mixture stays distinctly basic. The sodium picramate end product is itself a pH indicator which will provide a shift from an
intense dye dark red color to a nearly colorless supernatant liquid when the completed and diluted and filtered hot reaction mixture is acidified to
precipitate the free and nearly insoluble dull orange brown color picramic acid.
It would also seem possible that a lesser amount of ferrous sulfate could be used if dextrose or fructose along with the needed amount of NaOH to
enable its use as a reducing agent was used in substitution for the omitted quantity of the ferrous sulfate and NaOH, since the reducing sugar itself
will accomplish the reduction of sodium picrate to sodium picramate directly, or will act indirectly and in parallel to reduce the ferric hydroxide
byproduct back to ferrous hydroxide which should react again with the unreacted sodium picrate as a reducing agent regenerated by the reducing sugar.
Likewise should occur for regeneration of the ferrous hydroxide regardless of its source being sulfate or chloride or acetate, it should be
regenerated by basified glucose being freshly added to the reaction mixture.
The earlier estimation for a possible requirement of 9 moles of ferrous sulfate for the reduction of 1 mole of sodium picrate was uncertainty about
the mechanism that would predominate for the reduction. There is a Schikorr reaction
which is 3 Fe(OH)2 → Fe3O4 + H2 + 2 H2O
http://en.wikipedia.org/wiki/Schikorr_reaction
To supply the 6 hydrogens needed for the reduction by this Schikorr mechanism would require 9 moles total of the ferrous sulfate. And the byproduct
would be the mixed ferrous-ferric oxide Fe3O4.
[Edited on 28-3-2014 by Rosco Bodine]
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
You managed to dig up quite a bit of material actually, thanks. Once I get this term of school out of the way I may do a little experimenting. I am
starting to like DDNP, but the mono-reduction of picric acid is definitely a big source of inefficiency and inconvenience at the moment. Hope you’re
on to something good there.
Next time I prepare sodium hydrosulfide I am going to use a much larger excess of hydrogen sulphide gas. The unconverted sodium hydroxide causes low
yield and increased amounts of by-product formation.
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
I don't think the NaHS is the issue causing your reduced yields or that using an excess of H2S will be the fix. IIRC correctly the reduction reaction
is actually higher yielding for the Na2S used as the reductant instead of the acid salt NaHS which would incline your reduced yields more correctly
attributable to having too much H2S instead of not enough. You may be correct about the proportion of H2S to NaOH being the issue but are wrong about
the direction of the "sulfur value" involved disproportion with respect to the base that is causing the reduced yield. You are "over correcting" in
the opposite and incorrect direction IMO to use more H2S.
If I had to guess, the reduction of sodium picrate using sodium ascorbate it likely going to be the best method.
[Edited on 28-3-2014 by Rosco Bodine]
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
That was what I was trying to say in the last couple of posts, that it was most likely the larger proportion and concentration of sodium hydroxide
present in the reaction mixture causing the reduced yields. That was much earlier in the thread when I was thinking that I wasn't using enough
reducer. I may still not be using enough reducer, as well, but at this point I think the excess sodium hydroxide is the big issue. I am quite
confident that I can raise the yields considerably now that I understand what is happening (I think).
I don't think any extra sodium hydrosulfide is going to reduce the picramic acid further once formed. I do think that before the picramic acid is
formed it is susceptible to conversion to undesirable by-products especially in the presence of excess sodium hydroxide.
The excess hydrogen sulfide gas would be bubbled through the sodium hydroxide solution, when preparing the sulfide solution, in order to be more sure
that all the sodium hydroxide was converted to sodium sulfide or sodium hydrosulfide. I may also start using a little less sodium hydroxide when
converting picric acid to sodium picrate (use just enough). The paper mentioned a couple posts back talks about using sodium bicarbonate as a buffer
as well, with reasonably good results in the reduction reaction.
[Edited on 29-3-2014 by Hennig Brand]
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
No you are still not understanding. The Na2S forms first when you are gassing NaOH with H2S and is converted to the acid salt NaHS by further
saturation with H2S but the NaHS can be reverted back to Na2S by adding the needed Na equivalent as NaOH or NaHCO3. It isn't at all an excess of base
that is reducing your yields IMO it is a deficiency of base for the NaHS being used for the reduction instead of Na2S being used. The reduction of
picric acid to picramic acid is a misleading way of characterizing the reduction which is actually a reduction under basic conditions of a picrate
salt to a picramate salt. What exactly is the pH range that is limiting for the monoreduction, I am not certain, but even under basic condition it is
possible to go further than the monoreduction and form some byproduct diaminonitrophenol. This can occur even during reduction using glucose and NaOH
when the reducing agent is in excess of the amount needed for the monoreduction. So the maximizing of the yield for the monoreduction will be
dependant upon pH and time of reaction and temperature and the amount of excess of theory of reducing agent that is present and the concentration of
the reaction mixture. All of these parameters will have to be fine tuned to maximize the yield. There are some additional references I will be
posting.
It is odd that during the testing of reduction by glucose that the molar ratio used would not include the theoretical 6 moles glucose, and a bit in
excess of theory like 6.5 or 7, but instead the charting of the reduction would jump from 5 moles to 10 moles, leaving everyone to wonder what occurs
for the reaction dynamic in the near vicinity of the theoretical.
Attachment: The Alkaline Reduction of Aromatic Nitro Compounds with Glucose.pdf (286kB)
This file has been downloaded 713 times
Attachment: Effect of Glucose on Alkaline Picrate.pdf (846kB)
This file has been downloaded 851 times
Attachment: Reduction of Nitrocompounds by Sodium Ascorbate.pdf (155kB)
This file has been downloaded 842 times
Attachment: Basic Ferrous Salts journal article.pdf (87kB)
This file has been downloaded 853 times
[Edited on 29-3-2014 by Rosco Bodine]
|
|
|
roXefeller
Hazard to Others
Posts: 463
Registered: 9-9-2013
Location: 13 Colonies
Member Is Offline
Mood: 220 221 whatever it takes
|
|
| Quote: Originally posted by Rosco Bodine |
If I had to guess, the reduction of sodium picrate using sodium ascorbate it likely going to be the best method.
[Edited on 28-3-2014 by Rosco Bodine] |
I could never get that to take. I used a clear solution of sodium ascorbate. I ran it at boiling but the ascorbate seemed to decompose into some
black mess at least it looked like that.
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
The "black mess" probably contained the desired sodium picramate product which you failed to recognize and then failed to isolate. It is the expected
result that the orange colored transparent sodium picrate solution becomes so darkly red tinted and opaque during the reduction that it literally
turns black, and looks like black ink, because the sodium picramate product is such an intense and dense dark red color dye that undiluted it appears
black. Diluted greatly the red color should appear. Or the moderately soluble red sodium salt can be converted by acidification to the free picramic
acid which is nearly insoluble and will precipitate from acidified solution leaving a barely tinted supernatant. Sodium Picramate is used commercially
as a hair dye to produce a similar effect to henna where a red staining is imparted to hair exposed to the sodium picrate used as a hair colorant.
What may be the ultimate yield possible for the reaction as a synthesis intended to produce and isolate as the end product picramic acid gotten by use
of sugar as the reducing agent is not known. All of the literature about the reaction is focused upon the reduction to picramate of a picrate by
reducing sugars as a determination of sugar concentration gauged by colorimetry since the picramate byproduct produced by oxidation of sugar by sodium
picramate is an intense red dye, and the oxidation of the sugar goes to completion in the presence of excess picrate. Logically it seems possible
that the same reaction could be retasked and optimized for conversion of the sodium picrate to sodium picramate, perhaps with some adjustment of the
proportions of reactants with respect to what would be present in amount of theory or in excess of theory being the sugar, to enable more complete
conversion of the picrate to picramate, without need of solving for an unknown that is the sugar content. The "sugar test" is likely possible to be
reconfigured and retasked to be a conversion of sodium picrate to sodium picramate as the object.
Sucrose is inverted to glucose and fructose after heating at near boiling for 10 minutes with picric acid where the pH of the picric acid catalyzes
the inversion similarly as does HCl or other acids. When saturated sodium carbonate is added in excess to the heated mixture, the sodium picrate
formed will begin to oxidize the reducing sugars, with the sodium picrate being reduced to sodium picramate in the reaction.
A "black mess" should likewise be produced by this method. How sweet it is or may be as a method of synthesis for sodium picramate remains to be seen.
Attachment: Sodium Picramate for Hair Colorant.pdf (252kB)
This file has been downloaded 1142 times
Attachment: Some Modifications of Picric Acid Method for Sugars IND43966901.pdf (380kB)
This file has been downloaded 1322 times
Attachment: The Estimation of Glucose in Biological Samples 0200263.pdf (187kB)
This file has been downloaded 872 times
Attachment: Pages from Journal_of_the_American_Chemical_Society pg 403.pdf (317kB)
This file has been downloaded 697 times
Attachment: Pages from The Journal of Biological Chemistry pg529.pdf (188kB)
This file has been downloaded 639 times
[Edited on 30-3-2014 by Rosco Bodine]
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
I really wasn't trying to make any statements about the relative reducing power of sodium sulfide versus sodium hydrosulfide. I was thinking about
excess sodium hydroxide floating around in the reaction mixture.
What is attached is the Hodgson and Ward paper, which has already been posted at least once. When I read their results, I see nothing to indicate that
using only sodium hydrosulfide and no sodium sulfide would result in the diminished yields that roXefeller and I are seeing. The yield using only
sodium hydrosulfide and an amount of sodium hydroxide needed to neutralize the picric acid produced a yield of 86% (Table I, Exp (3)). There were
higher yields, with the highest yield (96%) coming from an experiment where a reducing sulfide solution containing equimolar quantities of sodium
sulfide and sodium hydrosulfide as well as an amount of sodium bicarbonate were used (Table I, Exp (1)).
In table II there are a couple of experiments where a slight excess of sodium hydroxide is used, which doesn't appear to make a huge difference to the
yields (at least nothing to account for someone only getting a 40% yield). Table II experiments were performed in water not methanol however.
I know one probable reason why the last sodium (hydro)sulfide solution I prepared was less dark yellow. It was a double batch, with approximately the
same amount of air (oxygen) in the generator which was bubbled through the solution. It would make sense that less yellow polysulfides were formed
(per unit volume).
Something I am doing differently from the Hodgson and Ward procedures; I am letting the reaction drag on for 45 minutes sometimes, instead of the
10-15 minutes specified by them. The next time I do the experiment I am going to add the reducing agent much more quickly.
Attachment: Picramic Acid using Sodium (Hydro)Sulfide J[1]. Chem. Soc., 1945, 663 - 665,.pdf (344kB)
This file has been downloaded 739 times
[Edited on 31-3-2014 by Hennig Brand]
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
I think you are misreading what is an admittedly awkward, unclear and confusing description of the "crystallized" Na2S reducing agent A, which is made
in situ by treatment of NaHS with NaHCO3. It was me who posted that reference back on page 3. There was some discussion in following posts about the
"sulfur value" being adjustable. That is the process done reacting the NaHCO3 with NaHS to form the Na2S. My understanding of this to be the case is
based upon the mole ratios stated being the same for reducing reagent A and B which makes sense best to me if the reducing agent A is Na2S formed in
situ. If this is interpreted differently then the molar ratio for A would be ~4.6 to 1 and the molar ratio for B would be ~2.3 to 1. There is seeming
contradiction between the numbers and the text description. I have a reference request for an earlier article which may help resolve the discrepancy.
[Edited on 31-3-2014 by Rosco Bodine]
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
Ok, I see what you are talking about. I think they are less likely to have the masses wrong, since those would have been read right of the scales (no
chance of calculation error and less chance of mix-up). Also the number of moles to be concerned about is the number of moles of sulfide not sodium.
No matter how much sodium we add we still can't get more than 1 mol of sodium sulfide from 1 mole of sodium hydrosulfide. I have a feeling that 24g of
sodium sulfide is right, but instead of 0.1 moles beside it, it should have said 0.3 moles.
I took the following from the allreactions.com website. It might provide some insight.
SODIUM HYDROSULFIDE NaHS
Properties of sodium hydrosulfide NaHS:
White, in the liquid state - black. It melts without decomposition, on further heating decomposes. Readily soluble in cold water (hydrolysis on the
anion). Decomposed in the boiling solution. Neutralized by the alkali (as opposed to NH4HS). Attaches sulfur. A typical reducing agent, is oxidized by
atmospheric oxygen.
Obtaining sodium hydrosulfide NaHS:
2 Na + 2 H2S (saturated) = 2NaHS↓ + H2↑ (in benzene).
Na2S + H2S (saturated) = 2NaHS.
H2S + NaOH (diluted) = NaHS + H2O
H2S + 2 NaOH(conc.) = Na2S + 2H2O.
2 H2S + 2Na = 2NaHS + H2 (150°C)
H2S + M(C2H5O) = MHS↓ + C2H5OH (M = Li, Na, K; in ether)
Reactions with sodium hydrosulfide NaHS:
NaHS = Na2S + H2S (450—500°C)
NaHS·nH2O↓ ↔ NaHS (saturated) + nH2O (normal temp., n >= 3).
NaHS (diluted) + 4 H2О (cold) = [Na(H2O)4]+ + HS-
HS- + H2O ↔ H2S + OH-; pKo = 7,02.
2 NaHS (solution) = Na2S + H2S↑ (boiling).
NaHS + НСl (diluted) = NaCl + H2S↑,
NaHS + 3HNO3 (conc.) = S↓ + 2 NO2 ↑ + NaNO3 + 2H2O.
NaHS + NaОН (conc.) = Na2S + H2O.
2n NaHS (solid) + (n - 1)O2 = 2 H2O + (2n - 4) NaOH + 2 Na2(Sn) [100—250° С].
NaHS (saturated, hot) + NaOH + (n - 1)S = Na2(Sn) + H2O
[Edited on 31-3-2014 by Hennig Brand]
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
It is a convoluted imprecise description article that could have been written better, and it becomes obvious there is error for example 6 in table 1
where the ratio should be 1.15 to 1 and it looks like a transposing error from example 4 above in the same table. The arithmetic I did showed a ratio
of 2.273 to 1 for what is shown on the table as 2.3 to 1 rounded. Anyway I think the patent I posted earlier about the reactions involved when
gassing NaOH with H2S is absolutely correct that the Na2S forms first and this would square with other process chemistry studies which have shown the
trend of higher sulfides forming first preferentially and then the sulfur value lowering as forced by conditions favoring change to what would
ordinarily be a less stable sulfur compound under ordinary conditions. The sulfur compounds have varying stability particularly for solutions which
are subject to instability after bottling and what begins as a pure solution of one sulfur compound gradually changes to an equilibrium mixture of
various sulfur compounds and can even precipitate sulfur in the process of reaching an equilibrium. The composition of the mixtures that are formed
is algebra that is affected by time and temperature. I was trying to get a handle on this to guesstimate what may be the best strategy for
sequencing a dry method reaction of solid NaOH and solid sulfur with minimal water and calculating the dilution and temperature and time for reaction,
sequenced with subsequent additions of added base as hydroxide or bicarbonate or carbonate as would produce an optimized mixture of reducing sulfur
compounds for the Zinin reduction, where the byproduct would be soluble thiosulfate and no precipitation or minimal precipitation of free sulfur as
byproduct would occur, and no sulfur dyes as byproduct with picramate would be an issue. I still think there is an optimization possible for such a
sulfide route, but the algebra for achieving it would likely come down to trial and error. The math only is helpful so far, and it complicates things
when the math reported by others is wrong for typos or editing errors, leaving a mystery to figure out. That's what we have here.
The appeal of the sulfide reduction is the concentrated reaction mixture and the rapidity and high yields possible at 98% or possibly even the
quantitative yield is attainable.
And I think high yields are possible even using dry method produced polysulfide Zinin reagents which are not reported so far as I know except by me
And maybe my math is not so good sometimes. I shall continue to scratch my
head and think on this some more. But there are possible also interactions with manganous and ferrous salt used concurrently with sulfide, and
variants that suggest quantitative yields are possible with the ferrous salt. The reference I posted regarding the formation of basic ferrous salts I
think provides information which may be useful ultimately in a strategy to utilize the ferrous salt as a regenerable reducing agent, possibly
regenerated even by sugar. Avoiding the sulfide entirely even though it is efficient is desirable since it seems to be a "nasty" synthesis leaving
some impurity even in those procedures where a high yield is produced. A cleaner and more pure product that could still be easily synthesized is what
motivates examination of the alternative methods.
Sulfur chemistry is a journey into the tall grass While on safari there,
probably good to bring a gun and matches. 
[Edited on 31-3-2014 by Rosco Bodine]
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
"It is a convoluted imprecise description article that could have been written better". I agree, there is a lot of vagueness and ambiguity in that
article. I found myself reading over parts of it several times and still being unable to tell exactly what the intended message was (and that was
before you pointed out the inconsistencies with some of the numbers).
What comes first seems to depend on the concentrations, or maybe the intermediate doesn't stick around very long. It is sort of complicated.
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Here are some additional references that may help untangling the mystery. The page 78 from the fundamental processes of dye chemistry is the one cited
by Hodgson and Ward, but a revised later edition is also attached since the language differs slightly or the later edition of the same article. From
what I can gather it is possible to improve upon yields which are obtained by optimizing the conditions with regards to temperature and time of
reaction and pH and the excess of the reducing agent employed. Evidently there are a number of different Zinin reagents or combination schemes that
are workable, as well as other non-Zinin reducing agents which can also give good yields, and the findings of different authors may generally agree on
some things and diverge on others. So the literature has to be read in a kind of selective way where the reporting on optimizing of one method does
not necessarily rule out good yields being also possible by a different scheme which may be rejected as infierior by the same author who never had
much luck with a different method which wasn't extensively studied or refined to see if it could be made to work. My own thinking is there are
probably many different reduction schemes that can produce a high yield of 80% or better and it isn't a reduction limited to only being practical by
one method which outshines all others. The hydrosulfide or the normal sulfide or the disulfide are all effective as reducing agents for the Zinin
reaction, but there are different requirements for pH control depending on which one or what mixture is used for the reduction.
Attached also is the referenced article: The monoreduction of 1,3-dinitronaphthalene and the separation of 3-nitro-1-naphthylamine and
4-nitro-2-naphthylamine,
Herbert H. Hodgson and Stanley Birtwell, J. Chem. Soc., 1944, 75-77
Attachment: Communications.pdf (375kB)
This file has been downloaded 962 times
Attachment: Pages from The_Fundamental_Processes_of_Dye_Chemistry 1921 pg78.pdf (84kB)
This file has been downloaded 788 times
Attachment: Pages from fundamental_processes_of_dye_chemistry.pdf (172kB)
This file has been downloaded 717 times
Attachment: Herbert H. Hodgson and Stanley Birtwell, J. Chem. Soc., 1944, 75-77.pdf (317kB)
This file has been downloaded 1083 times
[Edited on 2-4-2014 by Rosco Bodine]
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
The first attachment "Communications” is a very good article, thanks for posting. The conclusions they came to ring true for me, though they went
into more depth than I did with regards to theory. I think when bubbling hydrogen sulfide into a dilute sodium hydroxide solution, sodium hydrosulfide
is formed without going through sodium sulfide. Either that or the sodium sulfide is a very short lived intermediate. Sodium hydrosulfide is formed by
the half neutralization of hydrogen sulfide. I also think that a large excess of hydrogen sulphide gas must be bubbled through the solution, with the
simple setup used, or else all the sodium hydroxide will not be converted. I think that the excess sodium hydroxide causes undesirable reactions and
loss of yield. The next time I prepare sodium hydrosulfide I am going to be much more careful and use a large excess of hydrogen sulfide.
[Edited on 3-4-2014 by Hennig Brand]
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
There is a difference in the pH which is needed for the Zinin reaction to be favored depending upon the sulfur value or sulfur ranking of the sulfide
employed, and yields can be optimized accordingly by varying the reaction conditions to accomodate the sulfide being used. It is necessary for that
to be done so that apples and apples are being compared, rather than apples and oranges, when evaluating the "best obtainable yield" for a particular
sulfide being employed for the reduction. The solvent system and the concentration and temperature and time of reaction are also factors along with
the pH. There would need to be a broader more extensive study done to fairly compare the results gotten for use of NaHS and Na2S and Na2S2 or for
complex polysulfide mixtures, with and without the concurrent use of ferrous and manganous salts, and I think the use of schemes involving two phase
reaction schemes with organic solvents immiscible or partly miscible with the aqueous phase. There has likely already been proprietary studies done
by industry since this is an old compound having been manufactured on a plant scale for more than a century.
There are general principles for optimizing Zinin reductions which are in general most kinetically favored for the disulfide reagent. However, there
are exceptions to the general rule where one particular sulfide may be preferable as the anomaly for a particular reduction. Also the purity and the
byproducts may differ for different choices and govern the choice for what process is most preferred, so that absolute maximum yield is a lesser
priority, and other factors govern the choice of method. Reductions conducted under basic conditions produce a soluble thiosulfate byproduct, but for
reductions approaching neutral or acidic conditions, free sulfur will be produced as a byproduct that can complicate purification of the desired
product. So the economics of the process chosen will vary depending upon the requirements applicable to the quality of the desired product and its
practical use as an intermediate for further reactions, and the purity requirement applicable there. For a dye intermediate there may be one
specification, and for other uses a different specification may require a different process be used for producing a different specification end
product.
As a note of interest, I was reading a description of Na2S-9H2O as being a *white* coarsely crystalline material having the appearance of rock salt,
and being highly soluble like NaOH.
With regards to your H2S generation and saturating of an NaOH solution to produce the NaHS, there is a good measured indication of reaction progress
and endpoint result gotten by weight monitoring of the reaction flask as described in the articles. The scales won't lie to you about what you've
got. Possibly if you mean to maximize the yield, to increase 90% to 96%, instead of the NaHS being what you desire to produce by treatment of NaOH
solution with H2S, that instead you may perform that reaction only to the midpoint, so that your solution contains the normal sulfide Na2S, then
evaporate to obtain the crystallized hydrate. The added work involved may be poor economy so it is possible that the easier method producing 90% yield
from an aqueous system is more desirable than the alternative method used to increase yield at the expense of more work.
Looking at the Hodgson and Ward article in light of the Hodgson and Birtwell article about the interactions of Na2S and NaHCO3 in methyl alcohol
appears to be a scheme for improvising NaHS in situ which is the inverse of what I had guessed they were doing from the confusing description. I was
on the right track but going the wrong direction, so it was actually the purpose of mixing Na2S and NaHCO3 in methanol to make NaHS by an alternative
method to the separate synthesis of NaHS from a treatment of an NaOH solution with H2S from a gas generator. The Communications by Clayton article
uses NaHS gotten from treatment of NaOH solution with H2S. But the Hodgson - Birtwell and Hodgson - Ward variation uses alternative method of
producing the NaHS in methanol. Actually what I believe is achieved could be a hybrid method where a simultaneous Zinin reaction is proceeding where
in part the reducing sulfide is NaHS and in part the reducing sulfide is Na2S and in what exact proportions is unknown. The hybrid scheme could
actually serve to aid pH control by a kind of buffering effect, but I will have to again check the reaction schemes difference for NaHS and Na2S to
evaluate that aspect. Evidently an effect of that nature is involved because of the yield increase from 90% to 98%, although the solvent methanol
could also have bearing to an unknown extent. Further thought and analysis about this aspect is needed to fully understand the implications and what
accounts for the 6-8% differing yields. Actually that range of difference is small enough to be within the range of variability that depends on how
much skill by different chemsists is employed in conducting the synthetic process and manipulations. An under 10% difference range is within the
variation range of tweaking and fine tuning so what may be significant as actual chemistry differences becomes less conspicuous.
[Edited on 2-4-2014 by Rosco Bodine]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
| Quote: Originally posted by Hennig Brand | | The first attachment "Communications” is a very good article, thanks for posting. The conclusions they came to ring true for me, though they went
into more depth than I did with regards to theory. I think when bubbling hydrogen sulfide into a dilute sodium hydroxide solution, sodium hydrosulfide
is formed without going through sodium sulfide. Either that or the sodium sulfide is a very short lived intermediate. Sodium hydrosulfide is formed by
the half neutralization of hydrogen sulfide. I also think that a large excess of hydrogen sulphide gas must be bubbled through the solution, with the
simple setup used, or else all the sodium hydroxide will not be converted. I think that the excess sodium hydroxide causes undesirable reactions and
loss of yield. The next time I prepare sodium hydrosulfide I am going to be much more careful and use a large excess of hydrogen sulfide.
|
Yes I agree the Clayton article Communications does pretty well specify conditions that would be useful for a scheme where the NaHS Zinin reagent is
prepared by use of a H2S gas generator to treat an NaOH solution until weight increase indicates conversion to NaHS. This is a parallel type of gas /
liquid phase reaction scheme as is used for producing sodium hypochlorite or sodium bicarbonate, monitoring the completion point by weight and
possibly by the increasing blowthrough that is unreacted and reaches the scrubber. Bubblers are good for visual flow monitors for these type
reactions. And having the reaction flask sitting on a scale for weight increase monitoring is greatly helpful.
Reviewing this thread I see that 7 years ago I was already getting a pretty good handle on the approaches that could be workable for a laboratory
scale, and had an understanding of the milder conditions that would be provided by using H2S generation for producing either Na2S or NaHS. I
described this method for Na2S on page 4
http://www.sciencemadness.org/talk/viewthread.php?tid=433&am...
And the last couple of references clear up any misinterpretation about the composition of the "hybrid mixture" of Na2S and NaHCO3 used to increase the
yields to nearly quantitative. There is a buffering scheme at work there I think where the same equilibrium mixture of NaHS and Na2S could be made
using the H2S gas generation scheme, but interrupting the conversion of the initially formed Na2S before it is completely converted to NaHS by
measured weight gain. If you follow what I am describing, it would be even easier to make the hybrid reagent if the conversion of the initially
formed Na2S was stopped when
approximately two thirds of the Na2S had been converted to NaHS. The amount of NaHCO3 to be added to the methanol and sodium picrate aqueous mixture
would be reduced by the same two thirds as that amount would not have been needed for conversion of Na2S to NaHS in methanol, having been accomplished
by variation of the composition of the hybrid mixture done deliberately by modulating the H2S flow to the Na2S solution formed initially.
Such a modification of "incomplete conversion" of the Na2S to NaHS by interrupting the H2S gas flow, combined with adding methanol to the
aqueous sodium picrate reaction mixture, could increase the yields of 90% reported by Clayton using NaHS in the Communications article, to the 98%
yields reported by Hodgson and Ward using the equilibrium "hybrid" mixture of NaHS and Na2S made in situ in methanol by adding NaHCO3 to Na2S. Such
approach should shorten the time required for working the H2S gas generator and would be convenient to interrupt at a stage while it was still freely
absorbing in the still reactive Na2S not yet nearly completed conversion mixture. The only modifications otherwise would be the reduction in needed
NaHCO3 and the addition of methanol, to emulate the higher yielding reaction conditions described by Hodgson and Ward.
So I think that surveying the available literature with better comprehension in light of the most recently obtained references, allows for
proposing such a modified scheme to improve the yields to nearly quantitative.
Such a scheme of using the H2S gas generator is the most technically precise method of performing this type reaction. And this is likely the
approach that will produce the highest yields. However I still believe it is possible to further optimize the "dry method" of mixing solid NaOH and S
and reacting with added heat and a sequence of manipulations that can provide a useful Zinin reducing agent of generally predictable composition as
the result, if it is desired to avoid a scheme requiring use if a H2S gas generator. There would be an extensive experimental workup required for
study and charting results of such a scheme that would be good to know as an alternative method, and what level of efficiency to which that less
technically superior method is limited. Reduced yields may be acceptable for an improvised scheme where the additional equipment needed for the best
method is not available or is inconvenient.
As an additional reference that further describes some of the reactions and crystallization aspects related to isolation of Na2S I am attaching
US3666410 for its information value.
Science Madness has done it again here I think with a more comprehensive review of the literature regarding an obscure subject, researched over a
period of many years to arrive at a good general picture of the chemistry involved. Google sure knows who we are when it comes to certain information
and discussion of historically obscure and technically interesting topics for which compendiums of good information are simply otherwise unavailable
Call this thread a keeper.
Attachment: US3666410 Na2S Crystallization Process.pdf (578kB)
This file has been downloaded 652 times
[Edited on 3-4-2014 by Rosco Bodine]
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
Ok, I see that I have been a little confused about a few things. I have thought some more on this. Sodium hydrosulfide is formed first, but when there
is an excess of sodium hydroxide the neutralization doesn't stop at the half neutralization point (sodium hydrosulfide), but goes straight on through
to sodium sulfide. As the sodium hydroxide is used up (neutralized) by the incoming hydrogen sulfide gas stream the pH begins to drop and sodium
hydrosulfide begins to form until all the sodium hydroxide is used up and all the sodium sulfide is converted to sodium hydrosulfide. In order to
eliminate or convert all (or most) of the sodium hydroxide in solution all must be converted to sodium hydrosulfide. Keeping the sodium hydroxide
concentration very low is important to good yields when using the hydrosulfide solution for the mono-reduction of picric acid.
The pKa values for H2S and HS- are 7.0 and 7.2 respectively, which are very close together. Since they are so close together the protons in
both cases are reacted at close to the same pH. Sodium hydrosulfide is formed first but in the presence of excess sodium hydroxide goes directly to
sodium sulfide. As the sodium hydroxide is used up the equilibrium conditions are such that sodium hydrosulfide is the only end product. This is what
I think is going on.
[Edited on 3-4-2014 by Hennig Brand]
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
The relationship of Na2S and NaHS is the same as the relationship of Na2CO3 and NaHCO3 or Na2SO4 and NaHSO4 ......NaHS is the "bi" or acid salt as
opposed to the neutral salt. Na2S is the neutral salt. NaHS is the acid salt, which could also be called Sodium acid sulfide, or perhaps sodium
bisulfide, or sodium hydrosulfide. Sodium Sulfide and Sodium Hydrosulfide are not chemicals as frequently and commonly encountered as more familiar
salts. Therefore it isn't immediately recognized as would be the case for salts like the carbonates or bicarbonates particularly.
Time for a song or two illustrating "aspect ratios"
<iframe sandbox width="640" height="360" src="//www.youtube.com/embed/78O6--THTF0?rel=0" frameborder="0" allowfullscreen></iframe>
<iframe sandbox width="640" height="480" src="//www.youtube.com/embed/GfJWqjoekow?rel=0" frameborder="0" allowfullscreen></iframe>
[Edited on 3-4-2014 by Rosco Bodine]
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
Sodium Hydrosulfide and Sodium Sulfide Equilibrium
Took the following from the text "Textile Dyeing and Coloration". It discusses the sodium hydrosulfide and sodium sulfide equilibrium. My
understanding is still a little sloppy, since this subject is a little confusing and my mental resources are stretched pretty thin lately.
I am still looking for a pdf of the book, but for now I just took print screen shots and made a pdf out of them.
Attachment: Sodium Hydrosulfide and Sodium Sulfide Equilibrium - From Textile Dyeing and Coloration Text.pdf (331kB)
This file has been downloaded 1442 times
[Edited on 3-4-2014 by Hennig Brand]
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
My guesstimate of two thirds conversion of the Na2S appears to be in the ball park. That was just off the top of my head and I didn't check. But
evidently it got your attention what I recognized about that equilibrium. Since we are breaking new ground at least on the record, discussing the
finer points now being characterized about the difference between 90% yield of Clayton compared with 98% yield of Hodgson and Ward using a buffered
reagent, it is down to proof by experiment what actually occurs there. But I think this aspect involving the buffering effect of the equilibrium
mixture does nail it down as what is defining the different yields, and possibly the methanol is a smaller factor. But it could be both factors
involved that explains the different yields. Anyway this pins it down more precisely what is the difference there than anything i know about that is
published. But the techs who actually run the commercial processes have surely already identified these variables, if they were looking and trying to
fine tune their processes.
Actually based on the description of the equilibrium shifted so far in hydolysis as that reference indicates, my guesstimate is low regarding the
needed conversion of Na2S which should probably be better around 90-95% and with the sodium bicarbonate reduced accordingly. I have the right idea
but didn't go far enough on that first guesstimate. So if you actually do try the hybrid mixture I would revise my estimate to a 90% conversion of
the Na2S to NaHS as a good starting point for experiments. Two subsequent experiments at 85% and 95% conversion should identify the direction up or
down needed for an optimization.
[Edited on 3-4-2014 by Rosco Bodine]
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
Don't know if this one was posted before. It has some nice graphs and for a patent is an interesting, straightforward read.
Attachment: US2346550A Sodium Hydrosulfide Production Patent.pdf (377kB)
This file has been downloaded 672 times
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Yeah I posted that US2346550 patent earlier on the preceding page about 8 posts down. I have a screen grab of some text that is relevant too, but to
be honest I think we have already gone beyond the published literature specific on the topic of picramic acid with the collective analysis which we
have collected here in one place in this thread.
Attachment: Zinin related screenshot text.pdf (253kB)
This file has been downloaded 1033 times
Here are some additional references more general in nature to the Zinin reduction and the possible reactions, and are not articles specifically about
synthesis of picramate, but do provide more general insight about the 3 different reaction mechanisms that are possible depending upon the pH and the
sulfide or sulfides mixture being used.
Attachment: Reduction of p-nitrotoluene by aqueous ammonium sulfide.pdf (191kB)
This file has been downloaded 2989 times
Attachment: Kinetics of the Reduction of Nitrotoluenes by Aqueous Ammonium Sulfide.pdf (204kB)
This file has been downloaded 2008 times
Attachment: Kinetics of the Reduction of Nitrochlorobenzenes by Aqueous Ammonium Sulfide.pdf (590kB)
This file has been downloaded 859 times
Attachment: A Reduction of Nitrotoluenes by Aqueous Ammonium Sulfide Kinetic Study.pdf (557kB)
This file has been downloaded 1630 times
There is reported (pages from fundamental processes of dye chemistry) a need for simultaneously in a parallel stream adding HCl as a neutralizer for
the NaOH byproduct when using Na2S alone as the Zinin reagent but the addition of the HCl lags behind the gradual Na2S addition, so the reaction
system would be slightly basic. I think if you see the references I just posted above you will see that pH is going to affect the mechanism and that
the more nearly neutral reduction condition using only NaHS as the Zinin reagent is going to favor the mechanism producing free sulfur as a byproduct
rather than the soluble thiosulfate. So keeping the pH basic is going to make the reduction run cleaner by favoring the mechanism that produces the
thiosulfate byproduct. It may even be possible that the disulfide of polysulfide Na2S2 gotten by dissolving excess sulfur in the Na2S could
substitute for the use of the neutralizing HCl and favor a scheme where again the desired thiosulfate rather than free sulfur is the byproduct and
such variant could also be a high yield reaction. This is an aspect which causes me to have continued hope that a "dry method" or variation, easily
improvised Zinin reagent of specific proportions of NaOH and S that are reacted according to a specific heating and sequence of manipulation may still
be a workable method that avoids the H2S generation scheme.
[Edited on 4-4-2014 by Rosco Bodine]
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
It still seems logical that having the reducing agent being made up mostly of sodium hydrosulfide would be best. If you look at the reaction equation
where sodium sulfide is used there is a mole of sodium hydroxide formed for every mole of sodium sulfide consumed. This seems to be the reason that
sodium sulfide, when used by itself, results in such low yields. If sodium hydrosulfide is used as the reducing agent, the amount of sodium hydroxide
in the reaction mixture will be kept very low since the sodium hydrosulfide (acid salt) will neutralize, or mop up, the sodium hydroxide produced by
the reaction and form sodium sulfide in the process.
Still a little confused as to why sodium sulfide mixed with sodium hydrosulfide would actually produce higher yields than using just sodium
hydrosulfide by itself.
Reaction equation taken from Chinese explosives presentation slides attached.
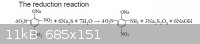
[Edited on 4-4-2014 by Hennig Brand]
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
| Pages:
1
..
5
6
7
8
9
10 |








