| Pages:
1
..
13
14
15
16 |
Lionel Spanner
Hazard to Others

Posts: 163
Registered: 14-12-2021
Location: near Barnsley, UK
Member Is Offline
|
|
Having made 7 attempts at it, 5 in tin cans and 2 in a stainless steel saucepan, I can only conclude the Grossmann method for making inorganic
nitrites (US Patent 792,515, 1905) which is cited in the wiki, is not useful or reliable. The results vary wildly depending on the surface material of
the reaction vessel, and when successful (in tin cans previously used for food) the nitrite was of very low purity, i.e. 40% or less. The two runs in
stainless steel produced no nitrite at all; the only product recovered was unreacted sodium nitrate.
|
|
|
Myc
Harmless
Posts: 7
Registered: 30-5-2022
Member Is Offline
|
|
I have been pursuing the old molten lead method over the last couple of weeks. 85 g Sodium nitrate and 207 g lead (I used lead fishing sinkers) were
melted in a cast iron pot and stirred continuously. An orange oxide begins forming, and the mixture bubbles. After about 30 minutes, the mixture is a
thick orange mud and the lead seems to have mostly reacted. Stirring is continued after the heat is stopped to prevent a solid mass forming. Once the
mixture solidifies, about 200ml water is added and the mixture left to soak for 15 minutes or so. All chunks should disintegrate.
The mixture is filtered and CO2 is bubbled through the filtrate for a few minutes; a white precipitate forms, which is removed by filtration. The
filtrate is then reduced by boiling. When the temperature reaches 125-130 degrees C, heat is removed and the mixture cooled to fridge temperature. The
crystals are then filtered and the filtrate reduced again for a second crop. Adding HCl to the salt gives plumes of brown gas. Pretty straightforward,
right?
Now it was clear to me from early on that not all the lead had reacted. There were small grains of metallic lead amongst the lead oxide. So I tested
the purity of my product as follows. 1 g of my salt was dissolved in a couple of ml of water. 2.5 g silver nitrate was similarly dissolved separately
in 5 ml or so. The two solutions were mixed; a white precipitate of silver nitrite formed instantaneously. This was filtered, dried and weighed. If
the 1g was pure sodium nitrite, the yield of silver nitrite should be 2.23g.
After one lead/nitrate reduction, my salt mixture was 30% nitrite. I then repeated the procedure with this salt mixture and fresh lead and got it to
50%. A third cycle got me to 80%. For reference I also tested some sodium nitrite isolated from curing salt, which I measured at 95% (although, in
this case, I think any sodium chloride contaminant would have formed the insoluble silver chloride which could throw my numbers off).
It's possible that a longer reaction could boost yield, however I'm also aware that sodium nitrite decomposes into the nitrate in the presence of
oxygen at high temperatures, so a longer reaction time could possibly be counterproductive. So all in all, to me, the lead method is 'not great'.
While I haven't tried it, the use of silver nitrate could also be used as a purification method if you're happy to buy/make plenty of silver nitrate
(you'll need 250g of silver nitrate to separate 100g of the sodium nitrite from nitrate contamination, but you can reclaim most of it!). After
reacting as above, the silver nitrite would be mixed with an equimolar amount of sodium chloride in solution to give a precipitate of silver chloride,
while sodium nitrite remains in solution. Filter the solid and evaporate the water for your sodium nitrite. The silver chloride can be made back into
metallic silver with NaOH and sugar, then reacted with nitric acid to regenerate the silver nitrate.
|
|
|
Lionel Spanner
Hazard to Others
Posts: 163
Registered: 14-12-2021
Location: near Barnsley, UK
Member Is Offline
|
|
For what it's worth I've recently tried a couple of methods described in this thread, with no joy.
The reaction of sulphamic acid, calcium oxide and sodium nitrate (as described on the first page) resulted in a lot of water vapour, some nitrogen dioxide, and the precipitation of a non-reducing substance that isn't nitrite
or nitrate, and is considerably less soluble in water than simple inorganic nitrites.
The reaction of sodium nitrate and sodium sulphide (as described in Morgan, 1908) produced sodium sulphate, and a hygyroscopic yellow product that was
slightly less soluble in water than nitrate or nitrite, and was contaminated with unreacted sodium sulphide - possibly sodium sulphamate.
Given that inorganic homebrewed nitrite is the modern-day amateur chemists' equivalent of the philosopher's stone, I'm very glad that Poland exists,
Polish vendors will freely sell sodium nitrite to private individuals, and although it's not cheap, the postage is not ridiculously expensive.
[Edited on 19-2-2023 by Lionel Spanner]
|
|
|
Lionel Spanner
Hazard to Others
Posts: 163
Registered: 14-12-2021
Location: near Barnsley, UK
Member Is Offline
|
|
Many years ago I used to work at a cosmetic/toiletry manufacturer that had once used bronopol (2-bromo-2-nitro-propane-1,3-diol) to preserve many of
its products, and found some of them turned brown to black due to the reaction between nitrite ions released by bronopol degradation and cocamide DEA,
turning the latter into an unstable N-nitrosamine due to an incomplete diazotisation reaction; when attempted with secondary amines, this reaction
stops at the nitrosamine intermediate. (As nitrosamines are highly carcinogenic, this was extremely bad news, and the preservative system in those
products was soon changed.)
As it turns out, bronopol is much more rapidly hydrolysed to nitrite in aqueous caustic soda at 100 °C. The initial products of the reaction,
formaldehyde and 2-bromo-2-nitroethanol, are relatively volatile, boiling at -19 and 83 °C. This could potentially be turned into a useful
preparation, though bronopol is hard to come by for amateurs.
Source: Sanyal, Basu, Banerjee. Rapid ultraviolet spectrophotometric determination of bronopol: application to raw material analysis and kinetic
studies of bronopol degradation. , J. Pharm. Biomed Anal., 14 (1996), 1447–1453. doi:10.1016/0731-7085(96)01779-7
|
|
|
Lionel Spanner
Hazard to Others
Posts: 163
Registered: 14-12-2021
Location: near Barnsley, UK
Member Is Offline
|
|
A major breakthrough!
One of the biggest problems in producing nitrite by reduction of nitrate is its tendency to react with oxygen at the reaction temperature. The
solution? Remove oxygen.
I recently got an argon cylinder over the counter from a local welding supply store (Machine Mart) and decided to retry the sodium sulphide reduction
method described in Morgan, in the same manner as the molten lead method (reductant added to molten nitrate in portions), while sparging the flask
with argon and keeping oxygen out.
It only went and bloody well worked!
I'll need to reproduce and refine the method before providing a full write-up, but this is definitely a viable way to produce nitrites at a small
scale. Get in!
|
|
|
clearly_not_atara
International Hazard
Posts: 2692
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
Chalk up another victory to the inert atmosphere! First eugenol demethylation, now nitrite.
Lovely choice of reducing agents we have. Sodium sulfide or lead. Any word on nickel carbonyl? 
But seriously, nice work!
[Edited on 04-20-1969 by clearly_not_atara]
|
|
|
Lionel Spanner
Hazard to Others
Posts: 163
Registered: 14-12-2021
Location: near Barnsley, UK
Member Is Offline
|
|
To be fair, sodium sulphide is easily obtainable from photography suppliers (it's the traditional reagent used for sepia toning), and you only need 1
mole of sulphide per 4 moles of nitrate. Plus, technical grade sodium sulphide is hydrated and is a liquid at the temperature the reaction is carried
out, so the reaction mixture is uniform, and you don't have the problem of uneven mixing that you'd get with a solid/liquid or solid/solid mixture.
The process described in Morgan was carried out in iron pans, with no mention of an inert atmosphere, which is a recipe for failure (and an awful lot
of ammonia.) The chemistry itself was sound, but the process was pretty bad.
|
|
|
Lionel Spanner
Hazard to Others
Posts: 163
Registered: 14-12-2021
Location: near Barnsley, UK
Member Is Offline
|
|
I couldn't reproduce this method.
In my initial attempt, I stopped when addition of sulphide started producing small deflagrations at the surface of the molten mixture, likely due to
formation of elemental sulphur - this started happening when 60% of the sulphide had been added. However, when I crystallised out nitrite, it was only
60% pure, suggesting an incomplete reaction had taken place. So the second time, I added all of the sulphide.
On dissolving the solidified reaction mixture, it appears the nitrite had been destroyed in the reaction, most likely due to sulphide reacting with
nitrite, forming elemental sulphur and ammonia; as no ammonia vapours were evident during the reaction, it was most likely captured as ammonium
sulphate. This would explain why although a highly soluble hygroscopic substance was produced on concentrating the mixture to near-dryness under
vacuum, it did not look like either sodium nitrate or nitrite (smaller crystals), and the pH of a solution was too low for it to be either nitrite or
nitrate (about 5).
Sod the shipping costs, going forward I'm just buying it from Poland.
|
|
|
fx-991ex
Hazard to Self
Posts: 67
Registered: 20-5-2023
Member Is Offline
|
|
In the first post he do NaHSO3 + CaCl2 and then heat the resulting Ca(HSO3)2 to decompose to CaSO3 + H2SO3.
I was thinking.
Lets start with sodium metabisulfite.
Na2S2O5 + H2O = NaHSO3
Then
NaHSO3 + Ca(OH)2 = CaSO3 + NaOH + H2O
Can this be done?(i know weak base displace stronger base, not the opposite, but the low solubility of CaSO3 will drive it forward?)
or (this should definitely work)
NaHSO3 + NaOH = Na2SO3 + H2O
followed by
Na2SO3 + CaCl2 = CaSO3 + NaCl
Also about the thermal reduction of NaNO3 + CaSO3, i plan to do it in a crucible(porcelain) with bunsen burner.
Do i heat it till the nitrate/nitrite melt?, is it gonna be hard to tell when it reached completion? or theres some color change?.
[Edited on 19-6-2023 by fx-991ex]
[Edited on 19-6-2023 by fx-991ex]
|
|
|
Lionel Spanner
Hazard to Others
Posts: 163
Registered: 14-12-2021
Location: near Barnsley, UK
Member Is Offline
|
|
Quote: Originally posted by fx-991ex  | In the first post he do NaHSO3 + CaCl2 and then heat the resulting Ca(HSO3)2 to decompose to CaSO3 + H2SO3.
I was thinking.
Lets start with sodium metabisulfite.
Na2S2O5 + H2O = NaHSO3
Then
NaHSO3 + Ca(OH)2 = CaSO3 + NaOH + H2O
Can this be done?(i know weak base displace stronger base, not the opposite, but the low solubility of CaSO3 will drive it forward?)
or (this should definitely work)
NaHSO3 + NaOH = Na2SO3 + H2O
followed by
Na2SO3 + CaCl2 = CaSO3 + NaCl
Also about the thermal reduction of NaNO3 + CaSO3, i plan to do it in a crucible(porcelain) with bunsen burner.
Do i heat it till the nitrate/nitrite melt?, is it gonna be hard to tell when it reached completion? or theres some color change?.
[Edited on 19-6-2023 by fx-991ex]
[Edited on 19-6-2023 by fx-991ex] |
My only comment is that you will need to work with an inert atmosphere, as nitrite salts are rapidly oxidised to nitrate by oxygen in their molten
state.
Good luck!
|
|
|
fx-991ex
Hazard to Self
Posts: 67
Registered: 20-5-2023
Member Is Offline
|
|
| Quote: Originally posted by Lionel Spanner | | Quote: Originally posted by fx-991ex | In the first post he do NaHSO3 + CaCl2 and then heat the resulting Ca(HSO3)2 to decompose to CaSO3 + H2SO3.
I was thinking.
Lets start with sodium metabisulfite.
Na2S2O5 + H2O = NaHSO3
Then
NaHSO3 + Ca(OH)2 = CaSO3 + NaOH + H2O
Can this be done?(i know weak base displace stronger base, not the opposite, but the low solubility of CaSO3 will drive it forward?)
or (this should definitely work)
NaHSO3 + NaOH = Na2SO3 + H2O
followed by
Na2SO3 + CaCl2 = CaSO3 + NaCl
Also about the thermal reduction of NaNO3 + CaSO3, i plan to do it in a crucible(porcelain) with bunsen burner.
Do i heat it till the nitrate/nitrite melt?, is it gonna be hard to tell when it reached completion? or theres some color change?.
[Edited on 19-6-2023 by fx-991ex]
[Edited on 19-6-2023 by fx-991ex] |
My only comment is that you will need to work with an inert atmosphere, as nitrite salts are rapidly oxidised to nitrate by oxygen in their molten
state.
Good luck! |
Apparently it can be done without melting the mixture (230C-300C)
Heres a video where it seem to be working well: https://www.youtube.com/watch?v=5BLPoE6Y-ns
|
|
|
Lionel Spanner
Hazard to Others
Posts: 163
Registered: 14-12-2021
Location: near Barnsley, UK
Member Is Offline
|
|
If you can actually get it to work, that'd be one hell of an achievement.
As I said: good luck!
|
|
|
Sir_Gawain
Hazard to Others
Posts: 268
Registered: 12-10-2022
Location: Due South of Due West
Member Is Offline
Mood: Yes.
|
|
Would it work better with potassium nitrate? KNO2 is 10 times more soluble than KNO3.
“Alchemy is trying to turn things yellow; chemistry is trying to avoid things turning yellow.” -Tom deP.
|
|
|
Lionel Spanner
Hazard to Others
Posts: 163
Registered: 14-12-2021
Location: near Barnsley, UK
Member Is Offline
|
|
Since reductive pathways to nitrite generally require high temperatures and produce poor purity products, or result in over-reduction to ammonia, it
may be worth taking a cue from the industrial synthesis, namely the reaction between nitric oxide and caustic soda, which doesn't involve any
oxidation or reduction at all.
Now nitric oxide itself is hard to come by for amateurs, but here's an idea for an indirect route: prepare nitrosyl sulphuric acid, dissolve it in
sulphuric acid, cool the solution to near 0 °C, then add it (slowly and carefully!) to an equally cold hydroxide solution, under an inert atmosphere,
to produce a mixture of nitrite and sulphate, that can easily be separated due to the large difference in solubility.
Brauer claims direct addition of water to nitrosyl sulphuric acid produces dinitrogen trioxide, a direct precursor to inorganic and organic nitrites,
but this is probably only true at temperatures well below zero.
Nitrosyl sulphuric acid is not trivial to make, as it requires sulphur dioxide to be bubbled through cold fuming nitric acid, but as a non-volatile
solid with a melting point of 70 °C it is easier to handle than other simple inorganic nitrosyl compounds, which are highly toxic gases or
low-boiling liquids.
[Edited on 2-8-2023 by Lionel Spanner]
|
|
|
Lionel Spanner
Hazard to Others
Posts: 163
Registered: 14-12-2021
Location: near Barnsley, UK
Member Is Offline
|
|
Here's what may be a very interesting and relevant paper, describing selective production of nitrite or nitrate by electrochemical oxidation of
ammonia with a copper electrode.
https://chemistry-europe.onlinelibrary.wiley.com/doi/epdf/10...
Unfortunately Sci-Hub hasn't indexed it, and I'm not in a position to drop the Britbongland equivalent of $49 just to download it, especially if it
turns out to be hot garbage. Would anyone who has access to the Wiley online library be able to share it? Many thanks!
|
|
|
Parakeet
Hazard to Self
Posts: 66
Registered: 22-12-2022
Location: Japan
Member Is Offline
Mood: V (V)
|
|
Here it is.
Haven't read everything yet, but the procedure looks amateur friendly.
Attachment: ChemSusChem - 2021 - Johnston.pdf (714kB)
This file has been downloaded 154 times
|
|
|
Lionel Spanner
Hazard to Others
Posts: 163
Registered: 14-12-2021
Location: near Barnsley, UK
Member Is Offline
|
|
Fantastic, thank you very much!
|
|
|
Fantasma4500
International Hazard
Posts: 1677
Registered: 12-12-2012
Location: Dysrope (aka europe)
Member Is Offline
Mood: dangerously practical
|
|
HNO3 --> isopropylnitrite!
i'd just like to report a pounding success. as im messing around with bulk dissolution of silver alloy in dilute nitric acid i showed off to one
non-chemist what nitric and copper can do, because its so toxic and beautiful, he had some acetone standing around and i added a bit into it, for
science- to see if anything interesting would happen. it died down and there was a vague organic nitrite scent, very mild vasodilatory effect, aha.
so i took a bit of copper wires, maybe 15% HNO3 and added about 1mL IPAlcohol to the.. 15mL mixture of acid
let it stand for maybe 3 hours roomtemperature, color changed very mildly due to copper metal dissolution
and, due to IPNitrite being very volatile i can indeed testify that theres a very strongly vasodilatory substance now in my 50mL erlenmeyer flask
so- simply scale this up, figure out how much % acid one can use, what temperature- preferably 50*C so you can immediatedly distill over the isopropyl
nitrite and right away react with sodium hydroxide to form our dear sodium nitrite in quite pure form
alternatively ethanol may be used, nitric acid and alcohol may cause a runoff and cause explosive formation thus its ideal to not increase the
temperature very much as for, distilling it out as its hard to tell what could set off organic explosives in a glass vessel, perhaps some solvent
could be used to extract the nitrites, but that would also risk carrying over an organic nitrate, which would then contaminate the nitrite
its plausible we can go as low tech as HCl + KNO3 + Cu + IPAlcohol --> IPNitrite + NaOH --> NaNO2 + IPAlcohol
update: i mixed 30mL mL IPA with 30mL 62% HNO3, and 90mL H2O, giving 120mL 15% HNO3
at room temperature this caused a slight runoff but clearly giving off some vasodilatory fumes, more dilution or controlling temperature better might
be the way to go, seems to work without copper, albeit more difficult to control
[Edited on 13-8-2023 by Fantasma4500]
|
|
|
unionised
International Hazard
Posts: 5102
Registered: 1-11-2003
Location: UK
Member Is Offline
Mood: No Mood
|
|
Thanks but...
"Large-scale nitrite production (NaNO2) is achieved by bubbling gaseous N2O and NO through a solution of NaOH and Na2CO3".
Really?
|
|
|
Parakeet
Hazard to Self
Posts: 66
Registered: 22-12-2022
Location: Japan
Member Is Offline
Mood: V (V)
|
|
| Quote: Originally posted by unionised |
Thanks but...
"Large-scale nitrite production (NaNO2) is achieved by bubbling gaseous N2O and NO through a solution of NaOH and Na2CO3".
Really? |
Yeah. I've also read the third reference. It should be NO2.
|
|
|
fx-991ex
Hazard to Self
Posts: 67
Registered: 20-5-2023
Member Is Offline
|
|
In water it is, but in Alcohol the sodium nitrite salt is more soluble, and since alcohol is easier to remove thats why i went with the sodium salt
instead of the potassium one.
I did try the reaction(didnt extract with alcohol yet)
When i mixed the sulfite and nitrate it was a bit endothermic, so it seem like it process very easily.
Also the sulfite/nitrate salt quantity on the video i posted are for the anhydrous sulfite salt, the user that made the video also made another video
on how he made the sulfite salt and i think he is using the dihydrate like i did so the ratio is wrong. Probably why he has low yield(except from the
fact he has lost a lot of product from overheating the mix and a broken beaker/kept the top part).
I think this method is very promising.
For 5G of NaNO3 its 9.187G of CaSO3.
$$Na2S2O5 + H2O \rightarrow NaHSO3$$ $$NaHSO3 + NaOH \rightarrow Na2SO3 + H2O$$ $$Na2SO3 + CaCl2 \rightarrow CaSO3 + NaCl$$ $$CaSO3 + NaNO3
\rightarrow CaSO4 + NaNO2$$ last one use heat - temp 230-300 C for 30-10 min.
[Edited on 14-8-2023 by fx-991ex]
|
|
|
Alkoholvergiftung
Hazard to Others
Posts: 151
Registered: 12-7-2018
Member Is Offline
|
|
I ve never tried this way but i think its for bigger scale lab. Production the best methode. You dont need to melt something and you can make it in
big Backers.
50 Parts Sodiumnitrate desolved in 150 Parts of Water. Add 350ml Ammonia solution with specific wight 0,96. After this give slowly 60 parts Zinc
powder to it and hold temperature between 20C and 25C.After an short time all nitrate is converted to nitrite.
Its from an patent who improved an old lab methode from Poggendorfs Analen of Physic and Chemistrie.
Concentrations and Temperatures are improtand if it isnt exactly followed the nitrate gets reduced to hydroxide.
|
|
|
Fantasma4500
International Hazard
Posts: 1677
Registered: 12-12-2012
Location: Dysrope (aka europe)
Member Is Offline
Mood: dangerously practical
|
|
Aluminium nitrite
aluminium metal doesnt react with nitric acid, and neither nitric acid vapors, but it will react with nitrate salts such as copper nitrate and iron
nitrate- however if aluminium can be kept seperate from a nitric acid / nitrate salt mixture the NO2 may react rapidly with the aluminium to form
aluminium nitrite
ideally the container is first flushed with CO2, butane maybe- as that is a very heavy gas
i did one attempt but the nitric acid/NOx fumes ate through the perforated plastic bag that was holding the shredded aluminium foil
(al foil flitter made by coffee grinding aluminium foil balls)
NO2 source was nitric acid and steel wool balls
i had a slight success proven by wettening the resulting mixture in IPAlcohol and adding dilute HCl which then formed IPNitrite, flame test faintly
showed IPNitrite but it was largely yellow, probably due to hydrogen effervescence and presence of sodium nitrate as i used Na2CO3 + HNO3 to form the
CO2 before i started the reaction, i shall attempt this further as this was a very poor attempt.
sadly it doesnt appear that aluminium carbonate exists, it decomposes into aluminium hydroxide which is difficult to filter as its a gel rather,
IPNitrite might be best way to scavenge the nitrite from this reaction
|
|
|
unionised
International Hazard
Posts: 5102
Registered: 1-11-2003
Location: UK
Member Is Offline
Mood: No Mood
|
|
Am I reading this correctly? The yield is about 50 micro moles per square cm per week.
So, with a 10cm by 10 cm electrode you would get less than a quarter of a gram of nitrite per week.
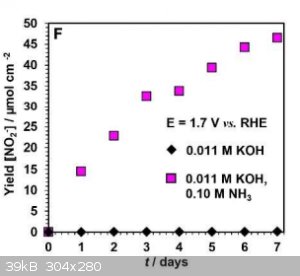
|
|
|
Kloberth
Harmless
Posts: 29
Registered: 2-6-2023
Member Is Offline
|
|
| Quote: Originally posted by Alkoholvergiftung | I ve never tried this way but i think its for bigger scale lab. Production the best methode. You dont need to melt something and you can make it in
big Backers.
50 Parts Sodiumnitrate desolved in 150 Parts of Water. Add 350ml Ammonia solution with specific wight 0,96. After this give slowly 60 parts Zinc
powder to it and hold temperature between 20C and 25C.After an short time all nitrate is converted to nitrite.
Its from an patent who improved an old lab methode from Poggendorfs Analen of Physic and Chemistrie.
Concentrations and Temperatures are improtand if it isnt exactly followed the nitrate gets reduced to hydroxide. |
Do you have a link to this patent or pdf?
|
|
|
| Pages:
1
..
13
14
15
16 |









