| Pages:
1
2 |
DJF90
International Hazard

Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
Thanks gc, I now have something to look forward to reading next week. It's a shame I can't read german 
|
|
|
ADP
Hazard to Others
Posts: 120
Registered: 4-4-2005
Location: USA
Member Is Offline
Mood: Productive
|
|
| Quote: | Originally posted by garage chemist
In the meantime I have succeeded in making SO3 from NaHSO4.
It worked extremely well, I got 23,8g SO3 from 100g pool pH-minus.
It's simply a matter of enough heat.
From 680-880°C, plain sodium pyrosulfate (from NaHSO4 at 480°C) gives off all its SO3.
And the best part: not a bit of it is decomposed to SO2. Because decomposition of SO3 requires a catalyst, like for example iron compounds, which are
lacking here.
I just have to write a documentation.
Oleum has now become a simple OTC preparation.
[Edited on 22-3-2008 by garage chemist] |
EDIT: I found the writeup, very impressive! I have 400g of NaHSO4 and look forward to doing this as a project.
[Edited on 20-5-2008 by ADP]
|
|
|
garage chemist
chemical wizard
Posts: 1803
Registered: 16-8-2004
Location: Germany
Member Is Offline
Mood: No Mood
|
|
Yes, of course, it's in Prepublication- has been for a long time.
|
|
|
Pulverulescent
National Hazard
Posts: 793
Registered: 31-1-2008
Member Is Offline
Mood: Torn between two monikers ─ "hissingnoise" and the present incarnation!
|
|
| Quote: | Originally posted by ADP
| Quote: | Originally posted by garage chemist
In the meantime I have succeeded in making SO3 from NaHSO4.
It worked extremely well, I got 23,8g SO3 from 100g pool pH-minus. |
I have 400g of NaHSO4 and look forward to doing this as a project.
|
Without wishing to detract, in any way, from what must have been a pretty sustained effort, garage chemist, IMO, a yield of 23.8g from 100g looks
somewhat skimpy for the amount of work expended.
If ADP gets a corresponding yield from his 400g he'll end up with less than 100g of product for his pains.
And I know this could be the maximum obtainable under the conditions.
I've seen a patent fairly recently which stated "The electrolysis is stopped at the point concentration of ~98%, since proceeding past this point
would result in the production of oleum."
It's not quite verbatim, and the patentee could, let's face it, be mistaken, for all anyone knows, but if he's right, it could be another grail, if
that's not a contradiction.
'One slight problem, I can't for the life of me, find it, or remember
which friggin' site I found it on.
It's frustrating, but I'm still looking.
If anyone else has come across something similar, I'd be most
interested?
I can't actually set up any kind of electrolysis at the moment, but might be able to have a go in a few weeks.
In the meantime, I want to look at the, so far, fugitive patent to see how authoritative it looks as I only skimmed through it the one time I saw the
blasted thing.
(sigh)Someone else must have seen it!
P
|
|
|
alphacheese
Harmless
Posts: 9
Registered: 7-12-2007
Member Is Offline
Mood: No Mood
|
|
| Quote: | Originally posted by garage chemist
Yes, of course, it's in Prepublication- has been for a long time. |
Forgive my ignorance... but where exactly
is the write up? A link maybe?
|
|
|
garage chemist
chemical wizard
Posts: 1803
Registered: 16-8-2004
Location: Germany
Member Is Offline
Mood: No Mood
|
|
Go to prepublication, press Ctrl + F, enter hydrogen, there it is.
|
|
|
alphacheese
Harmless
Posts: 9
Registered: 7-12-2007
Member Is Offline
Mood: No Mood
|
|
Ahh, fein dank. I didn't exactly know what the prepublication was. Found it.
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
I have been off and on working on building a small electric furnace for the last several weeks so that I can produce SO3 among other
things. The furnace was made from kiln bricks (2.5 inches thick), angle iron (2" X 2"), an old clothes dryer heating element (ca. 15 gauge nichrome
wire) and an infinite switch from an old kitchen range for temperature control. The infinite switch was rated for 240V, but seems to work fine at
120V. It is the current rating, in this case, which is of greater importance I think. I have a decent digital thermocouple meter but the best probe I
have has insulation rated to only 500C. I have ordered a probe that is rated for up to 1250C, but I don't have it yet. The 4000-6000W at 240V dryer
heating element was reduced in length slightly (cut) and was run at 120V instead of 240V. The current to the element was measured as about 9.5A when
the infinite switch was in the on state. This means the furnace element is putting out about 1140W (P=IV). With the infinite switch turned to the
maximum setting and the cover on the furnace an internal air temperature of 500 Celsius could be reached in about 2 minutes. I didn't actually time
it, but the temperature rises very quickly. Until I have the higher temperature thermocouple probe I won't know exactly how high the temperature can
go. I used the following video as a guide to build my furnace, but I also read David J. Gingery's book, "LI'L BERTHA, A Compact Electric Resistance
Shop Furnace", which had in it a lot of useful information.
http://www.youtube.com/watch?v=en4yhzLuD9A
Below are pictures of a trial run. I did two trial runs and led the SO3 into a Florence flask which contained what was left in the bottom
of a bottle of drain cleaner sulfuric (91% H2SO4). I obviously still need to do some work on the collection part of the
apparatus, as some SO3 was escaping and water was undoubtedly getting in and being absorbed, but the furnace works wonderfully and produces
SO3 from NaHSO4 with ease. I have read enough to know that I need a moisture protection tube with anhydrous calcium chloride or
some other suitable desiccant in it.
The quartz tube that a glass blower made for me is 30mm id. I had the side arm made rather long so as to avoid troublesome connections and also to
act as an air condenser. About 130-140g of sodium bisulfate (pool pH down) was put into the tube for each run. It was just a trial run as I said
before, but after the first run the 91% sulfuric acid in the flask was ~94% and after the second ~97% as measured by titration. I didn't measure the
volume at the start because I was mostly focused on the furnace for the initial runs, but the final volume of 97% sulfuric acid was 166mL. This is
likely not the best use of SO3, since the acid could be brought up to at least 95% reasonable efficiently by distilling off water. I also have a much
bigger quartz tube on the way. I am trying for a 50mm tube, with a little greater length also, which would allow me to make about 3 times as much
SO3 per run.
Air temperature in the furnace was kept between 300 and 400C for about an hour where it seemed mostly water came over. Once the air temperature in the
kiln got up over 500C it was not long until much more dense white fumes appeared and the drops coming out of the side arm made hissing sounds that
could be heard from 50 feet away when they hit the lawn below. The drops instantly carbonized the lawn they fell on. This is when I moved the
collection flask into place and began collecting SO3.
      
[Edited on 20-8-2014 by Hennig Brand]
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Zyklon-A
International Hazard
Posts: 1547
Registered: 26-11-2013
Member Is Offline
Mood: Fluorine radical
|
|
Nice! Did you calculate the % yields?
Surly they could have been increased by using a ground glass joint to a RBF rather then a Florence flask.
I doubt there'd be any pressure buildup, as the SO3 would dissolve quickly into the sulfuric acid.
How much 91% sulfuric acid did you start with?
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
I didn't have it all thought through when I gave the specifications to the glass blower for the quartz tube. I think the larger tube I am about to
have made will have the same design though, just a larger reaction tube. I think the long side arm/air condenser was a good addition and that by the
time the vapor makes it to the collection flask it will have cooled enough that a Teflon bushing could be used to connect the side arm to the flask
and the vent line to the flask. According to "Small Scale Synthesis of Laboratory Reagents", by Leonid Lerner, the maximum distillate temperature is
about 340C, even when the melted reaction mixture is at 820C. I may do something similar to what I did here, http://www.sciencemadness.org/talk/viewthread.php?tid=15676&... , but with a moisture protection tube connected to the vent line.
It is the non-condensable gases (air), in the system, that would be most responsible for building up pressure if the system was sealed. Maybe it could
be sealed once it was up and running, but I would feel a lot better about it being vented especially given the fact that the SO3 and
sulfuric acid are pretty dangerous materials to have an accident with.
I actually didn't measure the sulfuric acid volume started with, which is a real shame. It would have taken me 10 or 15 minutes to go get a graduated
cylinder and I was mostly just keen to test out the furnace and associated equipment. It turned out that the apparatus works really well and it is
actually a trivial matter to produce SO3 with a bit of equipment by this method. I wish I had measured the sulfuric acid volume at the
start because it would have told us how much SO3 was absorbed as well as how much water was absorbed. I suppose I could always do another
run in the same way.
[Edited on 29-7-2014 by Hennig Brand]
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
garage chemist
chemical wizard
Posts: 1803
Registered: 16-8-2004
Location: Germany
Member Is Offline
Mood: No Mood
|
|
Well Done, Henning Brand. It's nice to see that my work is still an inspiration to members.
For your next batch, try to condense the SO3 directly instead of absorbing it in H2SO4. Do not use a CaCl2 tube, the SO3 vapors will violently react
with every bit of moisture that it already contains. Instead, tightly stopper the neck of the flask with glass wool stuffed around the tube from the
condenser. This prevents circulation of moist air into the receiver. Cool the receiver with ice, immersing the flask in it.
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
Thanks Garage Chemist, I will definitely use your suggestions on my next batch.
Edit:
Ok, I ran another batch. I used the suggestion above and put some glass wool around the end of the side arm/air condenser so that it fit reasonably
snuggly in the neck of the Florence flask. I left it a little looser than I probably should have, mostly because I was a little nervous of sealing the
system off too tightly. I also used ice around the Florence flask as suggested and didn't add the concentrated sulfuric acid until after all the
SO3 had been collected.
Included are a few pictures that I thought were interesting. The charge of sodium bisulfate containing feed used was 130g, which came in a 4kg pail as
pH down for swimming pools. After SO3 generation had gotten quite slow, and increasing the furnace temperature no longer seemed to help,
the flask was removed and covered tightly with a polyethylene plastic bag. It only took me a few minutes to go and come back with 100mL of sulfuric
acid, but in that time the SO3 vapors had turned the polyethylene cover completely black. The flask holding the SO3 was left in
the ice bath, while I went for sulfuric acid, so the vapor pressure would have been quite low. When the 100mL of sulfuric acid was poured into the
flask holding the SO3, the SO3 dissolved very quickly (approximately a few seconds) and the flask got hot to the point of being
almost uncomfortable to hold.
Yield
About one gram of acid was measured out for each titration using scales accurate to hundredths of a gram. A sodium hydroxide solution was first
standardized against a known sample of HCl. Phenolphthalein was used. Only one titration was performed for the sulfuric acid before SO3
addition and one for after. Performing more than one titration for each and taking an average could have increased accuracy, but I was very careful.
As stated before the scales used were only accurate to hundredths of a gram and the amount of acid used for each titration was about a gram. Ideally
much more accurate scales should be used, but a larger quantity of acid could be used per titration as well.
Volumes of acid were measured with a 250mL graduated cylinder. The sulfuric acid density table from Perry's was used.
Pre SO3 Addition: Titrated as 94.4% H2SO4
0.944(100mL)(1.832g/mL) = 172.94g H2SO4
(1-0.944)(100mL)(1.832g/mL) = 10.26g H2O
Post SO3 Addition: Titrated as 97.2% H2SO4
0.972(114mL)(1.8364g/mL) = 203.49g H2SO4
(1-0.972)(114mL)(1.8364g/mL) = 5.86g H2O
H2SO4 added = 203.49g - 172.94g = 30.55g
SO3 added = 30.55g (80.066g/mol / 98.079g/mol) = 24.94g
H2O added = 30.55g - 24.94g - (10.26 - 5.86) = 1.21g
Theoretical Yield
According to the MSDS for the pH minus product used its composition is 91.5-94.7% sodium bisulfate and 4.8-8% sodium sulfate.
For 130g of Feed (assuming 91.5% NaHSO4)
0.915 * 130g / 120.06g/mol (80.066g/mol)(1 SO3 / 2 NaHSO4) = 39.67g SO3
For 130g of Feed (assuming 94.7% NaHSO4)
0.947 * 130g / 120.06g/mol (80.066g/mol)(1 SO3 / 2 NaHSO4) = 41.05g SO3
Percentage Yield of SO3
Assuming 91.5% NaHSO4 Feed = 24.94g / 39.67g * 100% = 62.9%
Assuming 94.7% NaHSO4 Feed = 24.94g / 41.05g * 100% = 60.8%
There is a purity versus quantity trade-off here. If collection had been started earlier more SO3 would have been collected, but the
product would have contained a larger proportion of water. It looks as though the glass wool did a great job of keeping the water out of the
collection flask. I still noticed a bit of SO3 escaping from the flask, through the glass wool, but the wool could have been packed much
more tightly and a lot more ice could have been used around the receiver as well (I was low on ice).
      
Just to clarify, the orange/red color of the sulfuric acid, in the last picture, is due to the dye or other contaminants in the sulfuric acid drain
cleaner used. Pure, highly concentrated, sulfuric acid is a clear viscous liquid.
[Edited on 16-8-2014 by Hennig Brand]
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
I have a few more pictures which I thought might be appreciated by someone trying to follow the experiment.
Once the furnace cooled down the quartz tube was removed and the sodium sulfate residue/plug was easily removed by turning the tube upside down and
gently tapping it on a soft surface like wood. Sometimes the sodium sulfate plug sticks a bit, but pouring a little water into the tube and letting it
soak down past and around the plug frees the plug and allows it to be simply dumped out by inverting the tube.
 
Here are a couple pictures I found on my phone from when I was building the furnace. The first picture shows the frame just after the welding was
completed. The second picture was taken during the middle of heating element groove filing and grinding. A metal file and a rotary cutting bit and
cordless drill were used to make the element grooves in the kiln brick. The third picture shows the infinite switch that is used. It is the old
fashioned type dial controller for the burner on an electric cook stove. This infinite switch was for one of the large burners on an old stove.
  
The kiln bricks were expensive for me, mostly because I live in a lower populated area of the country and anything remotely specialized often needs to
be ordered. The box of 12 kiln bricks was about $40 and because they came from halfway across the country shipping was about $35. The steel was
purchased from a scrap yard, as new steel, for about $10. I removed about 8 infinite switches from old stoves and about 6 dryer elements from old
clothes dryers, which I was allowed to take free of charge from the same scrap yard. The element will need to be replaced from time to time,
especially if operating the furnace at very high temperatures (>1000C for instance), but they can easily be found free (old clothes dryers) and
with this design replacement is a fairly simple procedure.
Attached below is a wiring diagram for an infinite switch.
Attachment: 120 & 240V Infinite Switch Wiring Schematic.pdf (75kB)
This file has been downloaded 1144 times
[Edited on 1-8-2014 by Hennig Brand]
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
Attached is an article discussing the results of a study done on the thermal decomposition of sodium and potassium bisulfates. I have thrown out
potassium bisulfate, on several occasions, as it was considered a waste product left in the boiler after nitric acid was made from potassium nitrate
and concentrated sulfuric acid. Sodium bisulfate is readily available and reasonably inexpensive, but it is an advantage to know about other feed
sources.
Attachment: The Thermal Decomposition of Potassium and Sodium-Pyrosulfate.pdf (259kB)
This file has been downloaded 1594 times
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Oscilllator
National Hazard
Posts: 659
Registered: 8-10-2012
Location: The aqueous layer
Member Is Offline
Mood: No Mood
|
|
I notice that everybody who has tried the persulfate decomposition has used glass apparatus of one form or another. Is there any reason that you
couldn't use a stainless steel container for this? It should easily be able to withstand the temperatures involved, and the decomposition from the
metal walls of the container would be fairly minimal, I would have thought.
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
From what I have read the amount of materials that can stand up to sulfur trioxide as well as sulfuric acid, at the broad range of concentrations
encountered and at the very high temperatures involved, is very limited.
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
Several days ago I was running the furnace and producing SO3 when it got dark. I took a few pictures as it was getting dark, and after dark, which I
think are very beautiful. Attached are a few of the better ones.
       
[Edited on 21-12-2014 by Hennig Brand]
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
A Simple Char Test for Determining The Approximate Time Collection Should Start
According to "Small Scale Synthesis of Laboratory Reagents", from the Sulfur Trioxide and Oleum chapter summary:
"
• SO3/oleum is prepared by the pyrolysis of NaHSO4 to a maximum temperature of 820°C.
• A 500°C cut gives 86% oleum at 90% yield. A 580°C cut gives 94% oleum at 65% yield.
• 100% volatilization of H2SO4 from NaHSO4 shows that the residue is entirely Na2SO4.
• The reaction can be conducted in an unprotected quartz tube. Na2SO4 does not attack quartz at the reaction temperature.
"
From page 182 in the same chapter:
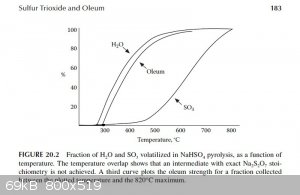
"Figure 20.2 shows data from the present experiment for the percentage of H2O and SO3 volatilized in a slow distillation as a
function of temperature. From this, it is seen that the separation of the two volatile components is reasonably efficient, so that at 500°C 90%
H2O and only 10% SO3 has volatilized, corresponding to an oleum strength of 86% free SO3 at 90% yield in the upper
cut. A cut at 580°C gives 94% oleum at 65% yield."
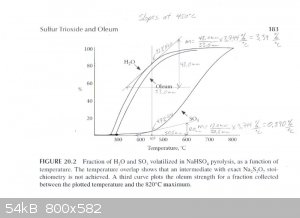
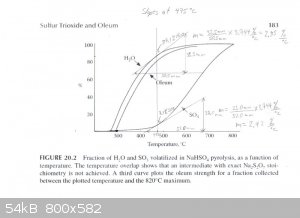
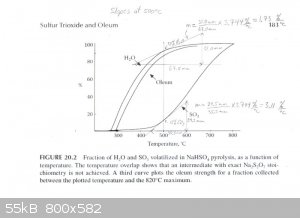
The graph described above was scanned and used to draw tangents to the water and SO3 curves at 450°C, 475°C and 500°C in order to
estimate the rate of water and SO3 production at those temperatures. From those rates the concentration of the product produced at those
temperatures was estimated. The graphs are attached. The following are the associated calculations that go with the graphs.
Basis: 1 mole SO3 & 1 mole H2O Produced in Total
Description of method used:
Instantaneous rates of change in terms of percent volatilized with respect to temperature were estimated by graphically determining the slope of the
lines tangent to the curves at each particular temperature. Those values were converted to moles per degree Celsius by assuming a basis of 1 mole of
water and 1 mole of sulfur trioxide produced. The flow rates found, for H2O and SO3, represent the compositions of the product
leaving the generator at the three temperatures examined. The x and y axis of the graph were found to be on different scales, so they were measured
and a factor calculated to relate the two.
(F (factor) = 3.744 %/°C)
At 450°C
H2O Instantaneous Rate of Change: (3.39%/°C) / 100 * 1 mole = 0.0339 moles/°C
SO3 Instantaneous Rate of Change: (0.890%/°C) / 100 * 1 mole = 0.0089 moles/°C
SO3 + H2O ----> H2SO4
0.0339 - 0.0089 = 0.025 moles of water left over & 0.0089 moles H2SO4 (anhydrous)
Total Mass flow = 0.025moles/°C * (18g/mol) + 0.0089moles/°C * (98.1g/mol) = 1.323g/°C
(0.873g/°C) / (1.323g/°C) * 100% = 66wt% H2SO4 Is Produced at 450°C
At 475°C
H2O Instantaneous Rate of Change: (2.85%/°C) / 100 * 1 mole = 0.0285 moles/°C
SO3 Instantaneous Rate of Change: (2.42%/°C) / 100 * 1 mole = 0.0242 moles/°C
SO3 + H2O -----> H2SO4
0.0285 - 0.0242 = 0.0043 moles/°C H2O & 0.0242 moles/°C H2SO4 (anhydrous)
Total Mass flow = 0.0043moles/°C * (18g/mol) + 0.0242moles/°C * (98.1g/mol) = 2.451g/°C
(2.374g/°C) / (2.451g/°C) * 100% = 97wt% H2SO4 Is Produced at 475°C
At 500°C
H2O Instantaneous Rate of Change: (1.73%/°C) / 100 * 1 mole = 0.0173 moles/°C
SO3 Instantaneous Rate of Change: (3.11%/°C) / 100 * 1 mole = 0.0311 moles/°C
SO3 + H2O ----> H2SO4
0.0311 - 0.0173 = 0.0138 moles of pure SO3 & 0.0173 moles of H2SO4 (anhydrous)
Mass Flow of SO3 = 0.0138 moles SO3 * (80.1g/mol) = 1.105g
Mass Flow of H2SO4 = 0.0173 moles H2SO4 * (98.1g/mol) = 1.697g
Product Concentration at 500°C = 1.105g / (1.105g + 1.697g) * 100% = 39% Oleum (well over 100% H2SO4)
What exactly is considered a good trade-off will of course depend on one's goals. My usual objective is to bring ca. 95% sulfuric acid up to 98-100%
sulfuric acid; so the goal is to raise the concentration while at the same time add as much weight of sulfuric acid as possible. From the data
obtained from the graphs, it is clear that starting collection when the acid produced by the generator is between 95-100% is a good trade-off in terms
of purity and yield, if increasing the concentration of concentrated sulfuric acid is the goal. When the generator is producing 97wt%
H2SO4, approximately 87% of the water has been removed and 93% of the SO3 remains to be collected. The concentration
increases very rapidly at this point, in relation to temperature, so even if collection is started at 95% H2SO4 it will be much
higher in short order. Using a char test is a simple way to determine when the acid concentration being produced is over 95%. It is quite easy to
determine the difference between 95% sulfuric acid and even 98% sulfuric acid by this method with a little experience. There is a huge difference in
the degree and rapidity of charing between 95% and 98% H2SO4 on wood or paper.
The Char Test Procedure
The following simple test has been used the last couple of times in order to determine the appropriate time to start collection:
Small pieces of scrap cedar have been used to determine when it was time to start collecting from the SO3 generator. A drop of 95% acid can
be placed on the test wood and watched to determine the rate and degree of charring. Once the furnace air temperature has risen to about 500°C the
output from the generator is periodically checked with the cedar block to see if the rate of charring is faster than the 95% reference. When the
output is determined to be over 95% sulfuric acid collection is started.
The yields obtained have been very noticeably better since following this new procedure. I will have to get back with a yield, when the generator is
run again, since the quick notes taken and measurement techniques used are insufficient to use to report an accurate yield. Also, without knowing the
exact temperature that collection was halted, in both cases, it is difficult to know exactly how much difference that may have made to the yields. In
any case, the ability to fairly accurately determine an appropriate time to start collection, without any special equipment, will improve yields and
reproducibility when producing SO3 by this method.
[Edited on 18-1-2015 by Hennig Brand]
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Microtek
National Hazard
Posts: 827
Registered: 23-9-2002
Member Is Offline
Mood: No Mood
|
|
I have been wondering about this for a while, after I saw a video of someone distilling SO3 from persulfate: It seems that in the persulfate reaction,
O2 is first liberated, generating pyrosulfate. Then when the temp is raised somewhat SO3 distills off.
It seems to me that apart from the initial loss of water/O2, the only real difference between these two procedures is that sulfuric acid is present in
the persulfate process, and that the temp is much lower.
So, has anyone tried heating NaHSO4 until, at some point, most of the water (and not so much of the SO3) has been removed, then adding some amount of
anhydrous H2SO4 to "liberate" the SO3 and distill it off at a relatively low temp?
It might be worth a shot if it hasn't been attempted already.
[Edited on 20-1-2015 by Microtek]
|
|
|
gdflp
Super Moderator
Posts: 1320
Registered: 14-2-2014
Location: NY, USA
Member Is Offline
Mood: Staring at code
|
|
There's another thread in Prepublication about SO3 from NaHSO4, not sure if that's what you're looking for though.
|
|
|
Microtek
National Hazard
Posts: 827
Registered: 23-9-2002
Member Is Offline
Mood: No Mood
|
|
I realise that this thread is called the persulfate method, but lately it has really been more about the bisulfate method. The point I'm trying to
bring up is the possibility of using sulfuric acid to (catalytically) liberate the SO3 from the pyrosulfate that is formed by removing a mole of water
from sodium bisulfate. There is indirect evidence that a lower temp may be required with this method, which could be interesting if true.
|
|
|
gdflp
Super Moderator
Posts: 1320
Registered: 14-2-2014
Location: NY, USA
Member Is Offline
Mood: Staring at code
|
|
Sorry, I misinterpreted your question. I don't see any reason why what you're proposing wouldn't work, you just need to get the bisulfate up to
around 400°C before it will dehydrate to pyrosulfate. Since some of it will decompose to sulfuric acid, you need a resistant container, but
borosilicate should handle those temperatures as long as it is heated reasonably slowly.
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
I see that I posted what I did in the wrong thread. I must have seen the bit of discussion on the bisulfate method in this thread and went from there.
Sorry about that.
Interesting idea Microtek. I know almost nothing about the persulfate method so I will have to read up, but I wonder if it is like the bisulfate
method in that there were assumptions made by many about the nature of the decomposition. Many believed that all or most water comes off at a more or
less well defined temperature and that the SO3 also comes off at a more or less defined, higher, temperature. In reality the water and SO3 are
liberated continuously throughout the temperature increase, but the rates change. Most of the SO3 comes off well above 500C, which is after the rate
of water evolution has dropped and SO3 evolution has increased to the point that only oleum is being produced. With the bisulfate method SO3 comes off
in high purity right up until 820C according to, "Small Scale Synthesis of Laboratory Reagents", which seems about right according to my own
experiments, whereas much of the literature indicates that SO3 is liberated sharply at a temperature around 450C. Would be interesting if it worked
though.
"A risk-free world is a very dull world, one from which we are apt to learn little of consequence." -Geerat Vermeij
|
|
|
Microtek
National Hazard
Posts: 827
Registered: 23-9-2002
Member Is Offline
Mood: No Mood
|
|
Very often in chemistry, the reality is less simple than the abstractions we use to convey the principle, so I'm not really surprised at the gradual
change in the composition of the distillate. I do remember someone posting a patent (article?) about using moderately reduced pressure (about 0.5 bar)
to effect the removal of water more cleanly.
As far as I'm concerned, the main attraction of this hypothetical procedure would be that it can be conducted in borosilicate glass instead of
expensive quartz.
|
|
|
XeonTheMGPony
International Hazard
Posts: 1636
Registered: 5-1-2016
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by Oscilllator  | | I notice that everybody who has tried the persulfate decomposition has used glass apparatus of one form or another. Is there any reason that you
couldn't use a stainless steel container for this? It should easily be able to withstand the temperatures involved, and the decomposition from the
metal walls of the container would be fairly minimal, I would have thought. |
Recently I been looking into this again and after some research carbon steal is compatible within acceptable limits to dry sulfur trioxide for the
discharge tube, the furnace tube will be consumed over time but wager you'd get a great deal worth of runs and given how cheap it will be!
Sulfate (Liquors) D-Severe Effect
Sulfur Chloride D-Severe Effect
Sulfur Dioxide D-Severe Effect
Sulfur Dioxide (dry) A-Excellent
Sulfur Hexafluoride N/A
Sulfur Trioxide C-Fair
Sulfur Trioxide (dry) A-Excellent
Sulfuric Acid (<10%) D-Severe Effect
Sulfuric Acid (10-75%) D-Severe Effect
Sulfuric Acid (75-100%) D-Severe Effect
Sulfuric Acid (cold concentrated) D-Severe Effect
Sulfuric Acid (hot concentrated) D-Severe Effect
Sulfurous Acid D-Severe Effect
Sulfuryl Chloride N/A
Source: https://www.calpaclab.com/carbon-steel-chemical-compatibilit...
So we can see the damage don to the reaction vessel is limited to the starting phase of the reaction, but I am willing to bet that with the lack of
moisture it should be minimal.
[Edited on 19-3-2017 by XeonTheMGPony]
|
|
|
| Pages:
1
2 |









