Electra
Hazard to Others

Posts: 179
Registered: 11-12-2013
Member Is Offline
Mood: No Mood
|
|
How do I best conduct a recrystallization when the impurities are similar solubilities as the product?
It's been a while since I've done any recrystallizations so I'm fuzzy on the theory.
For instance, say I alkylate a high molecular weight secondary amine with ethyl bromide, and the reaction proceeds to 90%, with the other 10% being
unreacted material. Both compounds would be very similar in their solubility in whatever solvent system is used, though I am unsure which is less
soluble than the other.
Would following a standard crystallization procedure work in this scenario? Dissolving it all, and then simply isolating the crystallized product,
leaving 10-15% of the mixture (mostly impurities?) in the solvent? Or is there a better methodology to approach this? I imagine the crystallization
could be conducted in either water with the salt form, or in a solvent like hexane with the base.
|
|
|
weilawei
Hazard to Others
Posts: 130
Registered: 3-12-2017
Member Is Offline
Mood: No Mood
|
|
A mixed solvent recrystallization might work here with a system like ethanol and water. It'll act more polar at high temperatures and less polar at
low temperatures. You ought to be able to get each product out individually with some tweaking (solvent choices, temperature ramp, etc).
|
|
|
Electra
Hazard to Others
Posts: 179
Registered: 11-12-2013
Member Is Offline
Mood: No Mood
|
|
I don't mind if I lose some of the unreacted material and the product, as long as I can isolate the the bulk of product in a relatively pure form.
|
|
|
SWIM
National Hazard
Posts: 970
Registered: 3-9-2017
Member Is Offline
|
|
Crystallization of a sample with that kind of initial purity (90%) often means (assuming it's done well) well-formed crystals which will tend to
exclude even slightly different molecules from their lattice.
This means you can often get purification that's a lot better than you might suspect from the chemical differences in the molecules.
This sort of situation calls for really slow crystallization that allows lots of time for the crystals to form.
Mixed solvent systems, as mentioned above, can be very helpful.
Best thing to do is to check out several organic lab manuals. Older ones that have experiments done on a regular scale and not these
ultramicro-thingies they give undergrads today.
These manuals will have instructions about doing such crystallizations, and choosing solvents and solvent mixtures for good results. Read a few and
give it a try.
It's nice when you can find detailed information on what solvents to use for workups of various reactions, but the principles used to choose solvents
you'll learn from these manuals will make you able to make better choices yourself when your dealing with 'unusual' products.
|
|
|
Electra
Hazard to Others
Posts: 179
Registered: 11-12-2013
Member Is Offline
Mood: No Mood
|
|
Thank you SWIM, for the info, specifically your first sentence gives me a lot of insight. I have read many different instructions and discourses on
recrystallization, in various online lab writeups, EDU pages, research articles, videos, and so on, but I've found very few of them go into much
detail about crystallization theory. I had been looking for ones that go into the more technical aspects of it, such as the theory you mentioned in
your first sentence about 90% being easily purified.
I realize the theory behind crystallization will differ greatly depending on what types of compounds are being worked with, and so far I've yet to
find an all encompassing guide.
|
|
|
RogueRose
International Hazard
Posts: 1585
Registered: 16-6-2014
Member Is Offline
|
|
I know that the speed at which the solution cools effects the size of the crystals, fast = small crystals, slow = larger. It does seem that there are
times when I've gotten large-ish crystals from putting a room temp solution in the freezer, getting fairly large "rock candy" like crystals - which I
would think is considered fairly fast cooling.
I was considering using a Thermos (glass, vacuum insulated & silver reflection inside) for recrystallizations as it should allow for a very slow
cooling of the solution.
I was also wondering how pressure or vacuum effects crystallizations. I would think that vacuum would lower the solubility and make more precipitate
- possibly fairly quickly and probably very dependent upon what is being crystallized. Increased pressure should increase solubility and help aid in
crystal growth, especially when heat is in the equation.
I'm wondering how slow the solution needs to cool for larger crystals to form (like rock candy, or min of 1-2mm in size). for the temp to fall 100C -
how long is the time period recommended? 2, 4, 8, 16, 24 hours?
|
|
|
unionised
International Hazard
Posts: 5102
Registered: 1-11-2003
Location: UK
Member Is Offline
Mood: No Mood
|
|
"How do I best conduct a recrystallization when the impurities are similar solubilities as the product?"
An answer would be to change the impurities into something with a different solubility.
Unfortunately, in this case it's tricky.
The best suggestion I could make would be to acetylate the mixture, then you can use the fact that amines are usually soluble in aqueous acid, but
amides are not, then you need to regenerate the product from the amide by hydrolysis.
It would work fine- as log as there was nothing "fragile" about the rest of the molecule.
A different approach depends on whether or not the two things you are trying to separate co-crystallise.
If you are lucky they don't. (It also depends on having "mainly" the stuff you want.
Imagine you have A and B which are both soluble at say 20% in hot water and 2% in cold water.
The mixture you have is 20 grams of a and 2 grams of B.
Heat them with 101 ml of water and the mixture all dissolves.
Then let it cool slowly.
The cold solution holds just over 2g of A and 2g of B
The crystals are (nearly) pure A.
A second crystallisation will lose you another 2g or so, but will give very near pure A
(If you are determined, at this stage you can repeat the reaction of the "leftovers" with ethyl bromide to make more stuff that's "mainly" the
material you want)
If you are unlucky, you get crystals that contain both solids, but that requires the two materials to pack into the same crystal lattice.
How will you know if you have a pure product?
|
|
|
yobbo II
National Hazard
Posts: 709
Registered: 28-3-2016
Member Is Offline
Mood: No Mood
|
|
Theory of fractional crystallization here. Not pretty reading.
How to Design Fractional Crystallization Processes
https://geocitieschloratesite.000webhostapp.com/chlorate/b_f...
|
|
|
Electra
Hazard to Others
Posts: 179
Registered: 11-12-2013
Member Is Offline
Mood: No Mood
|
|
unionised,
I was debating on taking the route of acylating the remaining secondary amine, or reacting it in some other way, but didn't think I had any reagents
for that on hand... As I was writing this post, I just remembered I have a little acetic anhydride laying around, so I might attempt to put that to
use. I'll probably do this route since I feel crystalization introduces too much chance that unreacted product will remain - which I must get rid of
almost completely or it would ruin the lithiation I want to do afterwards.
|
|
|
phlogiston
International Hazard
Posts: 1375
Registered: 26-4-2008
Location: Neon Thorium Erbium Lanthanum Neodymium Sulphur
Member Is Offline
Mood: pyrophoric
|
|
You mention acylating a secondary amine. Perhaps you can modulate the pH to affect the relative solubility of your precursor and product to achieve
separation?
-----
"If a rocket goes up, who cares where it comes down, that's not my concern said Wernher von Braun" - Tom Lehrer |
|
|
Electra
Hazard to Others
Posts: 179
Registered: 11-12-2013
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by phlogiston  | | You mention acylating a secondary amine. Perhaps you can modulate the pH to affect the relative solubility of your precursor and product to achieve
separation? |
After acylating the secondary amine (the impurity)? I'm not sure what you mean. If I were to acylate it, I would just do an acid base extraction since
the amide shouldn't form a salt. Or did you mean modulate the pH as a way to separate the secondary and tertiary amine in some way - which I don't
fully understand.
Part of my issue is that I lack analytical equipment, so with most separation methods, I have little way to verify the separation purity. With
something such as acylating the mixture, I can be pretty certain the secondary and tertiary are separated almost completely without much
post-verification.
|
|
|
DraconicAcid
International Hazard
Posts: 4278
Registered: 1-2-2013
Location: The tiniest college campus ever....
Member Is Offline
Mood: Semi-victorious.
|
|
You don't need to alkylate, acetylate or do anything fancy to separate the two. If it's 90% of the desired product, simple recrystallization should
do fine.
Consider compounds A and B, both of which have a solubility of 10 g/100 mL solvent at 90 oC and 1 g/100 mL solvent at 0 oC.
A mixture which contains 9 g A and 1 g B is added to 100 mL solvent and heated to 90 oC- all of it dissolves. We then cool it down to 0 oC. 1 g of
each compound stays in solution, so 8 g of A precipitates out, and all of the B stays in solution.
Bingo- pure A.
Crystallization will work even better if the impurities are more soluble than the product you're trying to purify. Since you want the compound with
the extra ethyl group (and one less N-H bond), you probably want to use a polar solvent such as an alcohol or mixed water-alcohol (assuming it will
dissolve in one of those).
Please remember: "Filtrate" is not a verb.
Write up your lab reports the way your instructor wants them, not the way your ex-instructor wants them.
|
|
|
Electra
Hazard to Others
Posts: 179
Registered: 11-12-2013
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by DraconicAcid | You don't need to alkylate, acetylate or do anything fancy to separate the two. If it's 90% of the desired product, simple recrystallization should
do fine.
Consider compounds A and B, both of which have a solubility of 10 g/100 mL solvent at 90 oC and 1 g/100 mL solvent at 0 oC.
A mixture which contains 9 g A and 1 g B is added to 100 mL solvent and heated to 90 oC- all of it dissolves. We then cool it down to 0 oC. 1 g of
each compound stays in solution, so 8 g of A precipitates out, and all of the B stays in solution.
. |
I understand the logic behind that, but what is the reason compound A/B stay in solution at a 1:1 ratio, when they were present at a 9:1 ratio total.
Is that just the magic of crystallization? s there some kind of formula at play here, similar to how fractions in fractional distillation work?
Btw I think you meant to say that 0.5g of each compound gets left in solution, not 1g of each. If it were 1g of each, wouldn't the solubility need to
be 2g/100ml @ 0c?
[Edited on 12-1-2018 by Electra]
|
|
|
DraconicAcid
International Hazard
Posts: 4278
Registered: 1-2-2013
Location: The tiniest college campus ever....
Member Is Offline
Mood: Semi-victorious.
|
|
| Quote: Originally posted by Electra |
I understand the logic behind that, but what is the reason compound A/B stay in solution at a 1:1 ratio, when they were present at a 9:1 ratio total.
Is that just the magic of crystallization? s there some kind of formula at play here, similar to how fractions in fractional distillation work?
Btw I think you meant to say that 0.5g of each compound gets left in solution, not 1g of each. If it were 1g of each, wouldn't the solubility need to
be 2g/100ml @ 0c? |
No, because the presence of A in solution will have a minimal effect on the solubility of B. The solubility of A in water at 0oC is 1g/100 mL, and
that is also its solubility in a saturated solution of B at 0oC (which is mostly water). If the solubility was higher, then things would be more
complex- A will be more soluble in a concentrated solution of B because they are chemically similar.
They stay in solution in a 1:1 ratio only because I handwaved the solubility and said they were equal. If the solubility of A was actually 0.8 g/100
mL at 0oC, then it would be a 0.8/1 ratio.
Please remember: "Filtrate" is not a verb.
Write up your lab reports the way your instructor wants them, not the way your ex-instructor wants them.
|
|
|
Electra
Hazard to Others
Posts: 179
Registered: 11-12-2013
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by DraconicAcid |
They stay in solution in a 1:1 ratio only because I handwaved the solubility and said they were equal. If the solubility of A was actually 0.8 g/100
mL at 0oC, then it would be a 0.8/1 ratio. |
So are you saying the ending ratio of the components in the solution post crystallization has nothing to do with their starting ratios in solution,
and only to do with their relative solubilities? So if I understand this correctly - if there were 7g of A, and 3g of B, with the equal aforementioned
solubility, would the result still be 1g of each left in solution, with the crystals containing 6g A and 2g B?
|
|
|
DraconicAcid
International Hazard
Posts: 4278
Registered: 1-2-2013
Location: The tiniest college campus ever....
Member Is Offline
Mood: Semi-victorious.
|
|
| Quote: Originally posted by Electra | | Quote: Originally posted by DraconicAcid |
They stay in solution in a 1:1 ratio only because I handwaved the solubility and said they were equal. If the solubility of A was actually 0.8 g/100
mL at 0oC, then it would be a 0.8/1 ratio. |
So are you saying the ending ratio of the components in the solution post crystallization has nothing to do with their starting ratios in solution,
and only to do with their relative solubilities? So if I understand this correctly - if there were 7g of A, and 3g of B, with the equal aforementioned
solubility, would the result still be 1g of each left in solution, with the crystals containing 6g A and 2g B? |
Exactly.
Please remember: "Filtrate" is not a verb.
Write up your lab reports the way your instructor wants them, not the way your ex-instructor wants them.
|
|
|
yobbo II
National Hazard
Posts: 709
Registered: 28-3-2016
Member Is Offline
Mood: No Mood
|
|
https://geocitieschloratesite.000webhostapp.com/chlorate/mut...
|
|
|
RogueRose
International Hazard
Posts: 1585
Registered: 16-6-2014
Member Is Offline
|
|
Well I thought I would do a test on how long some thermos's would retain their heat or how quick they would loose it.
I have 3 I'm testing a really cheap stainless steel inner wall (where liquid is in contact) and same as on outside). Then I have a VERY old Thermos
brand that is Al exterior (and cap), silvered glass and it uses an old cork to keep the liquids in - then an Al cap that screws on the top. the last
one looks kind of like a coffee pot that goes on a coffee maker, but it's a little different. It allows the coffee being maid (boiling) to drip down
into the "Thermos" and it is plastic with a silver reflective - glass internal and jas a screw on plastic lid that could be better IMO.
The containers were filled with hot tap water, allowed to sit for 30 mins, filled again, sat 2-3 mins, dumped, filled final time and measures starting
point temps.
The reason I'm doing this is because having a way to very slowly cool a solution may produce some really large crystals or at least some nice sized
ones while excluding a lot of the contaminates that may be in the mix.
I'm going to try this later with temps starting at 212F and see how long they take to get to ~60-70F (or maybe even down to 32F.
So if you want to try to use an insulated vessel like this you just have to make sure to be extremely careful upon removing the growing crystals from
the side of the glass because on the other side of the glass in under vacuum, so it can break easier (it's not super easy, just easier than normal, no
repeatedly tapping with a chisel to get a lump off the side wall)
I'm going to do 24 hours worth of data or so, unless some need more time to drop to room temp.
This is the image file of the spreadsheet hours 1 - 6.5
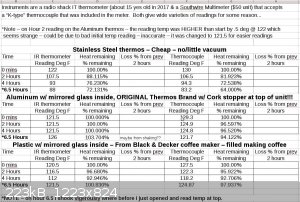
This is just the PDF of the spreadsheet hours 1-4
Attachment: Thermous Heat Retention over 24 hours - 3 thermouses - 2 temp sensors.pdf (34kB)
This file has been downloaded 274 times
[Edited on 1-14-2018 by RogueRose]
|
|
|
RogueRose
International Hazard
Posts: 1585
Registered: 16-6-2014
Member Is Offline
|
|
Well IOP let the thermos's sit over-night and was surprised at the results. room temp dropped from 72F to 60-62 for about 18 hours, so that should
have speed the cooling. The reading is at 30 hours from initial reading and 2 of these units, with the mirrored glass inside & vacuum (though
these are probably 30-50 years old, so IDK how good the vacuum is in them), had lost between 12.5% or 20% of the heat! That is pretty amazing in my
book and think these may work well for growing crystals slowly.
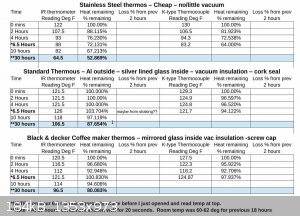
|
|
|
unionised
International Hazard
Posts: 5102
Registered: 1-11-2003
Location: UK
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by DraconicAcid | You don't need to alkylate, acetylate or do anything fancy to separate the two. If it's 90% of the desired product, simple recrystallization should
do fine.
Consider compounds A and B, both of which have a solubility of 10 g/100 mL solvent at 90 oC and 1 g/100 mL solvent at 0 oC.
A mixture which contains 9 g A and 1 g B is added to 100 mL solvent and heated to 90 oC- all of it dissolves. We then cool it down to 0 oC. 1 g of
each compound stays in solution, so 8 g of A precipitates out, and all of the B stays in solution.
Bingo- pure A.
Crystallization will work even better if the impurities are more soluble than the product you're trying to purify. Since you want the compound with
the extra ethyl group (and one less N-H bond), you probably want to use a polar solvent such as an alcohol or mixed water-alcohol (assuming it will
dissolve in one of those). |
Unless (as I said earlier) A and B co-crystallise.
Go on... try it with alum (white) and chrome alum (so dark it's nearly black).
You will get very pretty crystals of pale violet to deep amethyst.
And you will possibly get to learn that there are pitfalls to this sort of thing.
|
|
|
DraconicAcid
International Hazard
Posts: 4278
Registered: 1-2-2013
Location: The tiniest college campus ever....
Member Is Offline
Mood: Semi-victorious.
|
|
| Quote: Originally posted by unionised | Unless (as I said earlier) A and B co-crystallise.
Go on... try it with alum (white) and chrome alum (so dark it's nearly black).
You will get very pretty crystals of pale violet to deep amethyst.
And you will possibly get to learn that there are pitfalls to this sort of thing.
|
The OP was talking about two organic compounds, one of which had an NH group, the other had an N-Et group. The chances of them cocrystallizing are
far smaller than the chances of two ionic compounds with common ions and identical crystal habits doing so.
Please remember: "Filtrate" is not a verb.
Write up your lab reports the way your instructor wants them, not the way your ex-instructor wants them.
|
|
|








