DubaiAmateurRocketry
National Hazard

Posts: 841
Registered: 10-5-2013
Location: LA, CA, USA
Member Is Offline
Mood: In research
|
|
Thermocalculations in Energetic Materials
Introduction and Method
Recently, I visited almost every thermochemical calculation methods out there. Most of them are quantum mechanics - quantum chemistry based, while
some involves emprical fitting while anothers are group add:
CBS-X such as CBS-4M, CBS-QBS. G3, G3MP2, and G2. The W1 theory:W1U. B3LYP at 6-21, 6-31, 6-31g(d), 6-311g+(2d,p), and B3PW91 at 6-31g(d,p). PM3, PM6,
PM7 from MOPAC. Group adding from EMDB1.0 trial version, Keshavarz et al.
Equations that I have tried:
Ochterski 2000. [1]
Rice et al, 1999. [2]
Bryd & Rice 2006. [3]
Poltizer et al, 1997, 2001, and 2005. [4,5,6]
Klapotke, 2017 [7]
Here are some advantages and disadvantages of each that I have to give.
PM3, PM6, PM7 are fast and empirical-based methods. By fast I mean extremely fast, they're all almost below 1 minute. This is a method I want to talk
about a lot since it fits amateurs the most. Since they're fitted to empirical data, it gives rather far-off deviations toward novel structures.
TTTO's W1U (the absurdly accurate 0.3kcal deviation method) is 230.7 kcal, and all PM methods give off extremely far deviations toward this number
(>20kcal). The heat of formation of PM methods on furoxans, 1,2,4,5-tetrazines, 1,2,3,4-tetrazines, 1,2,3,4-tetrazine-2,4-dioxides, and other
similar N→O structures gives off high deviation. Their performance on nitro groups vary. PM7's usual deviation on energetic materials is around 6-12
kcal/mol.
B3LYP 6-31g(d) is the most computationally affordable method with relatively acceptable errors.
B3LYP 6-311g+(2d,p) is more accurate on tetrazine dioxides.
W1U method is affordable only on <20 atoms on a super-computer, but its result is probably more accurate than experimental ones.
CBS-QB3 and CBS-4M are relatively accurate but crash on large molecules.
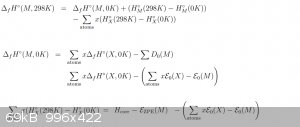
For most of the below, I use the above equation unless otherwise stated:
_______________________________________________________________________________________________
Results
PM3, PM6, PM7
Here is a list of PM3, PM6, PM7 results and their deviation:
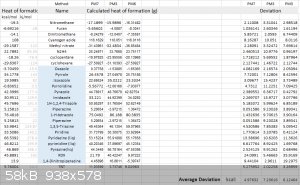
PM7
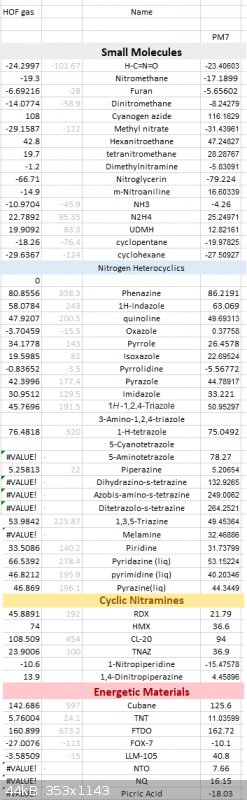
Here is a long list of PM7 results against various compounds, some of the nitrogen compound's gaseous HOF are calculated using ESP method against
their experimental data.
W1U
W1U method is the one of the most computationally expensive method. It is far out of reach for my computer to run anything bigger than methane, I
tried ethane and after half a day, it worked, but it will not run 1-H-tetrazole or triazoles. 
B3LYP 6-31g(d)
B3LYP-6-31g(d), Rather accurate average deviation here.
B3LYP 6-31g(d) 1999 Rice Correction
The same as above, however using a new equation fitted for energetic materials by Rice et al 1999.

B3LYP 6-311g+(d2,p)
B3LYP-6-311g+(2d,p) Its suppose to give better results than the one above, however did not, although I have not yet ran the entire test set that I
personally prefer yet. Some promising improves in tetrazine dioxides results are seen here.

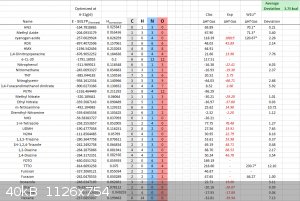
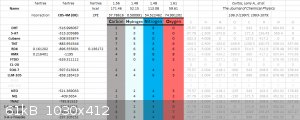
CBS-X
CBS-X method, I deleted a lot of data, so it looks unfinished, however the average error for solid state heat of formation was about 6kcal/mol for
CBS-4M, only 2 compounds were performed on CBS-QBS and CBS-APNO. For me at least, CBS-X methods run into errors on any compound bigger than TNT and
RDX. (12 hours of HMX and 20 hour CL-20 runs provides weird errors reports).
_______________________________________________________________________________________________
Sugestions and guide:
PM methods are available on the free program MOPAC, the installation procedure is difficult and is almost like an IQ test - I failed few times!
Carefully read citation [1]'s last few pages, they have literally all the numbers you need. EZPE is 0 for B3LYP based methods. All the results you
will obtain using these are gaseous heat of formation. For solid, use equations from either [2] or [6].
Multiwfn is a free software to obtain surface area and average electrostatic potential of a molecular surface. This is required for calculation of
ESP-method of enthalpy of sublimation, which is required for the solid phase heat of formation.
Gaussian 09 has all the basis set above. Potentially ORCA, PSI4, GAMESS, Spartan, and a few others have these too. ORCA and GAMESS is free. I have
personally also use ORCA for other calculations, however through cmd, and it took me a while to get used to it.
I never took chemistry and had to drop out of calculus II in college, there is some readings to do, but I believe anyone can do it  Obtaining Gaussian is hard, try asking forum members is the best legal tip I can
provide. Obtaining Gaussian is hard, try asking forum members is the best legal tip I can
provide.
_______________________________________________________________________________________________
Reference and Extra Reading:
[1] Ochterski, Joseph W. "Thermochemistry in gaussian." Gaussian Inc (2000): 1-19. http://gaussian.com/thermo/
[1] Rice, Betsy M., Sharmila V. Pai, and Jennifer Hare. "Predicting heats of formation of energetic materials using quantum mechanical calculations."
Combustion and flame 118.3 (1999): 445-458.
[2] Byrd, Edward FC, and Betsy M. Rice. "Improved prediction of heats of formation of energetic materials using quantum mechanical calculations." The
Journal of Physical Chemistry A 110.3 (2006): 1005-1013.
[4-6]
-Politzer, Peter, et al. "Calculation of heats of sublimation and solid phase heats of formation." Molecular Physics 91.5 (1997): 923-928.
-Politzer, Peter, et al. "Computational characterization of energetic materials." Journal of Molecular Structure: THEOCHEM 573.1-3 (2001): 1-10.
-Politzer, Peter, et al. "Computational prediction of standard gas, liquid, and solid‐phase heats of formation and heats of vaporization and
sublimation." International journal of quantum chemistry 105.4 (2005): 341-347.
[7] Klapötke, Thomas M. Chemistry of high-energy materials. Walter de Gruyter GmbH & Co KG, 2017.
Simmie, John M. "A database of formation enthalpies of nitrogen species by compound methods (CBS-QB3, CBS-APNO, G3, G4)." The Journal of Physical
Chemistry A 119.42 (2015): 10511-10526.
and its supplementary files: https://pubs.acs.org/doi/suppl/10.1021/acs.jpca.5b06054/supp...
Simmie, John M., and Kieran P. Somers. "Benchmarking compound methods (CBS-QB3, CBS-APNO, G3, G4, W1BD) against the active thermochemical tables: A
litmus test for cost-effective molecular formation enthalpies." The Journal of Physical Chemistry A 119.28 (2015): 7235-7246.
and its supplementary files:
https://pubs.acs.org/doi/suppl/10.1021/jp511403a
_______________________________________________________________________________________________
And Yes, I can calculate the theoretical heat of formation of novel compounds for you upon request under this post.
[Edited on 21-8-2018 by DubaiAmateurRocketry]
|
|
|
simply RED
Hazard to Others
Posts: 206
Registered: 18-8-2005
Location: noitacoL
Member Is Offline
Mood: booM
|
|
Really good work! Congratulations! Many thanks for sharing the results.
One thing to note, the energetic materials community works in MJ/kg. For example TNT E(detonation) is ~ 4 MJ/kg and that of RDX is ~ 5.5 MJ/kg. So it
could be nice the enthalpies of formation to be presented in MJ/kg also, much more intuitive.
Also, you may try to calculate some energies of detonation or combustion.
E(detonation) = E(molecule) - E(products)
Another one: ZPE = 0 is impossible! By 0 you mean you did not calculate it. Quantum oscillators have zero point energy even at 0 K, ZPE is never 0.
You calculate enthalpy of formation at 0 K and 298 K, do you know how the program does it? First it calculates the vibrational /and optionally
rotational/ states energies and then populates these energies to the point of 298 K. As you can imagine, this calculation is by itself ZPE corrected.
[Edited on 21-8-2018 by simply RED]
When logic and proportion have fallen sloppy dead...
|
|
|
DubaiAmateurRocketry
National Hazard
Posts: 841
Registered: 10-5-2013
Location: LA, CA, USA
Member Is Offline
Mood: In research
|
|
red, yes, it seems on B3LYP that the eZPE is already corrected, for some reason.
edit, otherwise, the equation would not work, this weird eZPE issue is same for B3LYP and CBS-x methods for anyone who need to calculate enthalpy.
[Edited on 21-8-2018 by DubaiAmateurRocketry]
|
|
|
simply RED
Hazard to Others
Posts: 206
Registered: 18-8-2005
Location: noitacoL
Member Is Offline
Mood: booM
|
|
If you perform vibrational analysis, the program may automatically correct ZPE. Whether or not, it should be written in the manual of the software.
When logic and proportion have fallen sloppy dead...
|
|
|
DubaiAmateurRocketry
National Hazard
Posts: 841
Registered: 10-5-2013
Location: LA, CA, USA
Member Is Offline
Mood: In research
|
|
So recently after a lot of effort to do computational chemistry for energetic materials, I can help anyone here to compute enthalpies to relatively
high accuracy.
I have access to both extremely high-end workstation computers as well as cluster computers. I am running Linux WebMO interface with Gaussian, NWChem,
ORCA, and MOPAC.
I also have a GTX1080Ti graphics card currently doing nothing on my shelf which i am looking forward to in GPU accelerating Gaus/NWChem calculations.
Methods such as CBS-4M, CBS-QBS, W1U (up to 10 atoms), G3MP2B3 can obtain results to extremely high accuracy (average deviation for G3MP2B3 is
2.5kcal/mol).
Currently, I'm working on crystal structure prediction, which seems to be an extremely hard task, I am looking at the software USPEX at the moment.
|
|
|
|










