hodges
National Hazard

Posts: 525
Registered: 17-12-2003
Location: Midwest
Member Is Offline
|
|
Non-Trivial DNA Experimentation at Home?
I read its pretty easy to extract DNA (see, for example, http://learn.genetics.utah.edu/units/activities/extraction/). But is it possible to do anything non-trivial with the extracted DNA? Such as (for
example) uniquely identifying an organism based on its DNA, introducing new genes (Recombinant DNA), etc.? Are biological reagents "watched" like
many other chemicals are? Can they be purchased by the science hobbyist?
I did a scan of messages in Biochemistry and don't see much on the topic here. Which leads me to think that either this type of experimentation
cannot be done very readily at home, or else maybe it is a bit off topic in a chemistry forum. Thoughts?
Thanks,
Hodges
|
|
|
12AX7
Post Harlot
Posts: 4803
Registered: 8-3-2005
Location: oscillating
Member Is Offline
Mood: informative
|
|
My (admittedly lacking) understanding is that it requires enzymes and whatnot in order to muck around with DNA. Reading (sequencing) it requires
extremely sensitive tests differentiating the segments, which can only be done for limited lengths of strings. Fortunately, enzymes and whatnot do
provide the mechanisms for these extremely sensitive tests, but it still must be carried out carefully and accurately.
Now, someone please do correct me. 
Tim
|
|
|
not_important
International Hazard
Posts: 3873
Registered: 21-7-2006
Member Is Offline
Mood: No Mood
|
|
Identification and gene tweaking are rather different, although the second generally uses the first as a tool.
There's a set of equipment needed that you're not likely to have, although at least some of it is within amateur DIY reach.
Identification can take many forms. Sequencing a lot of the DNA uses expensive equipment. Sequencing selected segments or mDNA is less daunting, but
still uses automated gear. DNA comparison, where a known and unknown DNA sample are compared is simpler, taking advantage than related but not
identical DNA strands will pair up to give DNA with a lower melting point than the pure samples - a match/no-match test.
Some of the off-the-shelf stuff around
http://www.zymoresearch.com/products/all_dna_products.asp
|
|
|
chemoleo
Biochemicus Energeticus
Posts: 3005
Registered: 23-7-2003
Location: England Germany
Member Is Offline
Mood: crystalline
|
|
Well, for DNA extraction, also see this thread https://sciencemadness.org/talk/viewthread.php?tid=1496
You can use papain (health stores) to digest/destroy protein, leaving you with DNA.
But, hodges, you are correct I'm afraid.
To do efficient manipulation of DNA you need 1) restriction enzymes (which is precisely what is used for DNA identification, or comparisons between
humans, etc (see RFLP analysis, http://en.wikipedia.org/wiki/RFLP) 2) PCR (and particularly the enzymes required for this), 3) Primers (short DNA segments that align with your
target sequence), and a staining compound such as ethidium bromide, agarose gels etc for visualising DNA.
It's actually really very much so easy to do once you have the reagents. I don't think these compounds are restricted, you could for instance always
contact New ENgland Biolabs, and see whether they sell restriction enzymes. Be prepared to pay. A lot. With this alone, you could do a genomic digest,
and compare the differences. All that's required at that point is a gel electrophoresis setup, and I'm sure you can improvise on the nature of the
gel.
As to isolation of restriction enzymes, please check http://en.wikipedia.org/wiki/Restriction_enzymes . You could try and isolate i.e. EcoR1, a restriction enzyme specific to the DNA sequence GAATTC,
from Ecoli (a restriction enzyme cuts DNA at a specific base pair sequence). This is relatively easy to grow, and I can help you on how to extract
total protein preparations. Then incubate with your DNA of choice, and compare the electrophoresis gels prior and post EcoR1 digest, which gives you a
unique RFLP imprint (i.e. DNA banding pattern on the electrophoresis agarose gel).
In fact, you may remember this thread, https://sciencemadness.org/talk/viewthread.php?tid=4380, about organisms growing at unusual conditions. I found something growing in our CuSO4
waterbath - where CuSO4 is meant to be toxic to fungi. I might subject this to precisely this treatment, or alternatively, get primers to clone up a
sequence within the mitochondria (I assume, this is fungal & eukaryotic). Sequence comparison with known sequences in the database should allow
one to identify the nature of the organism, or at least, to which organisms it is related to.
[Edited on 11-12-2007 by chemoleo]
Never Stop to Begin, and Never Begin to Stop...
Tolerance is good. But not with the intolerant! (Wilhelm Busch)
|
|
|
chemoleo
Biochemicus Energeticus
Posts: 3005
Registered: 23-7-2003
Location: England Germany
Member Is Offline
Mood: crystalline
|
|
DNA experiments with nucleases
You could also try experimentation along the lines of DNA nucleases (enzymes that non-specifically cut DNA at random positions).
For instance, extract DNA as described in the link above. Then, take a protein extract of pretty much whatever, and add a tiny quantity (like one
microliter) to your purified DNA. Watch the degradation of DNA with time, using agarose gels and the staining compound (there's one called GelStar
which might be easier to use and get).
YOu could even try to purify the DNA as whole chromosomes, or at least attached to nucleosomes (little protien units that wrap around DNA, and thereby
protect it from nucleases), and try the nuclease assay. Only exposed segments will be degraded. THis might lead to an organism-specific degradation
pattern too. If you want I can dig up material on this.
[Edited on 11-12-2007 by chemoleo]
Never Stop to Begin, and Never Begin to Stop...
Tolerance is good. But not with the intolerant! (Wilhelm Busch)
|
|
|
hodges
National Hazard
Posts: 525
Registered: 17-12-2003
Location: Midwest
Member Is Offline
|
|
Thanks for the replies. I think I will work on some electrophoresis experiments over the holidays (beginning first with simple dyes, possibly
progressing to DNA fragments) as a start. I found the following on one of the sites referenced in a DNA discussion thread here:
http://www.nexusresearchgroup.com/fun_science/electrophoresi...
Seems like a simple enough setup to create at home.
Hodges
|
|
|
chemoleo
Biochemicus Energeticus
Posts: 3005
Registered: 23-7-2003
Location: England Germany
Member Is Offline
Mood: crystalline
|
|
Wow, who would have thought that methylene blue,
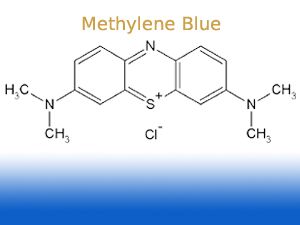
stains DNA - usually compounds such as ethidium bromide, an intercalator of DNA (it inserts into the helices of DNA and thereby changes fluorescence
exitation/emission)
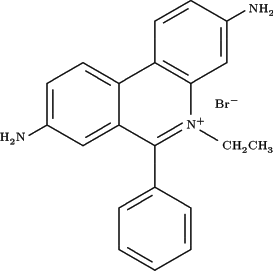
does so - I imagine it must have similar but weaker intercalating properties?
Regardless - if you can get hold of MB then a big hurdle is already overcome.
Do check this for details on concentrations and incubation times:
http://www.bio.net/bionet/mm/methods/1993-December/010023.ht...
| Quote: | # Load 2-5X the amount of DNA that would give bands of moderate intensity on an ethidium bromide stained gel. Typically this is something on the
order of 0.5-2.5 µg of a 1 kb fragment on a 30 ml 1% mini gel. These numbers are guess-timates so your milage may vary.
# Run the gel normally and then place in a 0.002% methylene blue (w/v, Sigma M-4159) solution in 0.1X TAE (0.004M Tris 0.0001 M EDTA) for 1-4 h at
room temp (22°C) or overnight at 4°C. Diffusion of the DNA does not seem to be a problem for fragments as small as 100 bp (3% Nusieve:1%agarose
gel). This avoids background issues associated with staining with 0.02% methylene blue for 30-60 min and then destaining for what seems to be forever
# If destaining is needed to increase the visibility of the bands place the gel in 0.1X TAE with gentle agitation changing the buffer every 30 - 60
min until you are satisfied with the degree of destaining. |
Never Stop to Begin, and Never Begin to Stop...
Tolerance is good. But not with the intolerant! (Wilhelm Busch)
|
|
|
hodges
National Hazard
Posts: 525
Registered: 17-12-2003
Location: Midwest
Member Is Offline
|
|
Electrophoresis
I performed electrophoresis using regular agar and a 30-volt power supply with nichrome wires at the edge of the agar dish for electrodes. Buffer
covering the agar was the usual weak NaHCO3 solution. I took the picture after removing the buffer, rinsing the agar, and placing into a regular bowl
for a better background.
The approximate run time was somewhere between one and two hours. It is hard to know exactly, because I was using thin nichrome wire and it kept
breaking (positive electrode) so I had to replace the wire several times. That is also probably why the bands moved slightly to the left, instead of
straight down. Sometimes the right part of the positive electrode broke off. I tried copper wire but it was worse than nichrome wire in that it
produced far more precipitate. The brown stain at the bottom of the plate is oxides/hydroxides of nickel and chromium from the positive electrode.
I ran a mixture of one drop food coloring, 5 drops water, and two drops glycerin for each sample. From left to right, the samples were red, green,
and blue.
Red appears to be a pure color. It can be seen that green is actually a mixture of blue and yellow. Blue appears to be a mixutre of purple and some
other light red color. Based on distances the bands moved, yellow is the lightest molecule, followed by red and then light red. Purple is the
heaviest.
(114 kb) http://www.sciencemadness.org/scipics/Electrophoresis.jpg
Now to find some DNA and a suitable stain.... If anyone knows a cheap source let me know. From science companies, DNA samples are around $100.
Hodges
|
|
|
Glucose Oxidase
Harmless
Posts: 37
Registered: 31-12-2012
Member Is Offline
Mood: Researching Alchemy
|
|
if you are interested in some DNA experimentation i am doing a research about protein synthesis by injecting its corresponding DNA into an E.Coli cell
any help will be extremely appreciated 
|
|
|
Thebrain
Harmless
Posts: 14
Registered: 26-5-2012
Member Is Offline
Mood: indignant
|
|
What is the state of kitchen counter or basement DNA manipulation these days? Does anyone know of successful genetic recombination outside of a
university of industrial laboratory?
|
|
|
phlogiston
International Hazard
Posts: 1376
Registered: 26-4-2008
Location: Neon Thorium Erbium Lanthanum Neodymium Sulphur
Member Is Offline
Mood: pyrophoric
|
|
Glucose oxidase, the basic principle is quite simple.
The most common method essentially consists of the following steps
(1) prepare a circular piece of DNA (called a 'plasmid') containing the gene coding for the desired protein, and a second gene which provides
resistance to an antibiotic.
(2) insert the plasmid into the bacteria.
(3) Grow the bacteria on a medium containing the antibiotic so that only the bacteria containing the DNA survive
(4) Grow larger amounts of the bacteria
(5) Harvest, break the bacteria and extract the desired protein.
There are many small details however that can make this an annoying chore for biochemists.
Most often, you just want to get as much of the protein as you can quickly, and in an active form, but in real life it typically takes a few weeks
from start to finish, and occasionally it may not work at all, for instance because the bacteria cannot be convinced to make a protein that is alien
to them, or they make it but fold it improperly so it aggregates or is inactive.
Making the DNA used to take most time, but these days you can buy ready-made plasmids that have been prepared by automated DNA synthesis machines for
very competetive pricing.
As for doing this at home: If you can get a plasmid I think it may be possible.
However, the DNA work is difficult to improvise. I agree that getting the enzymes and primers used to manipulate DNA will probably be the limiting
factor. Restriction enzymes, DNA polymerase, DNA ligase, etc. They are essential, and there is no easily improvised substitute I can think of.
[Edited on 25-1-2013 by phlogiston]
-----
"If a rocket goes up, who cares where it comes down, that's not my concern said Wernher von Braun" - Tom Lehrer |
|
|
Glucose Oxidase
Harmless
Posts: 37
Registered: 31-12-2012
Member Is Offline
Mood: Researching Alchemy
|
|
Thanks for the explanation phlogiston but i already have the resources to make a considerable amount of E.Coli and i researched a feasible injection
method (using CaCl2 and a heat shock "cold shock as a matter of fact" ) but yet my problem is getting a plasmid
though think i need a higher budget to be able to carry this out (i can get E.Coli for free & CaCl2 is toooooo cheap)
|
|
|
Tsjerk
International Hazard
Posts: 3022
Registered: 20-4-2005
Location: Netherlands
Member Is Offline
Mood: Mood
|
|
If you are really willing to do DNA work you could try to isolate the enzymes from E.coli. It is a lot of work but it is doable.
Nowadays laboratoria buy all their enzymes, but one of my teachers once told me how they did it about 30 years ago, they had to grow and extract their
enzymes, a multiple day job, to do a couple of reactions.
Restriction enzymes, DNA polymerase and DNA ligase are isolate-able, you only need an E.coli containing the protein expressing plasmid. As you are
already able to grow and transform E.coli you only need the plasmid. Maybe you can get it by contacting some research group or company producing the
enzymes... Once you have those you are in theory able to make any plasmid you want if you manage to get or make primers for PCR reactions (as long as
you have the organism containing the gene of interest). PCR can be done without a PCR machine as they did in the begin years, otherwise you can get
one for 300-400 dollars (new) or you can build your own. I once had a lecture from somebody who won some prices building a 100 dollar PCR using hear
dryer heating elements and such for diagnosing malaria in dark Africa, but I can't remember his name....
It is really die-hard molecular biology, which will be very time consuming, but in theory it is possible. Would be very cool to see someone pull this
of in their garage.
|
|
|
Thebrain
Harmless
Posts: 14
Registered: 26-5-2012
Member Is Offline
Mood: indignant
|
|
cool biohacking article in BBC
I found this article on the topic in the BBC a couple of weeks ago.
http://www.bbc.com/future/story/20130122-how-we-became-bioha...
|
|
|
White Yeti
National Hazard
Posts: 816
Registered: 20-7-2011
Location: Asperger's spectrum
Member Is Offline
Mood: delocalized
|
|
I remember making recombinant e-coli in biology in high school.
Not only do you need to trim the DNA with a nuclease, but you need to use the same exact nuclease for all cuts so that the "sticky
ends" latch onto each other properly. In addition to making DNA fragments, you need to insert a promoter and an operator to make a full operon.
Since the plasmids were already provided, we didn't have to do any of the delicate steps (thankfully). Even then, the "cold shock method" is pretty
unreliable, with low success rates. About a dozen to two dozen bacteria are actually genetically modified out of a zillion from the initial colony.
Genetically modifying mosses looks like a better avenue for biochemical synthesis en masse. It would be much more difficult than engineering
bacteria, but give it some time, and costs will come down.
"Ja, Kalzium, das ist alles!" -Otto Loewi
|
|
|












{kind=link}