| Pages:
1
2 |
Sauron
International Hazard

Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
CCl3Br -> CCl4 or CHCl3
I have just received a liter of trichlorobromomethane that I ordered some months ago
In this country carbon tetrachloride is verboten, and so is chloroform.
But they have overlooked CCl3Br which is perfectly importable without any special permit.
So what I am seeking is a way to replace the -Br with -Cl to obtain carbon tetrachloride, and/or replace the -Br with -H to obtain chloroform.
So far in the lit. I have found only studies of the ClBr interhalogen compound and really nothing that looks preparatively useful to me.
So I am soliciting help with this project. It seems to be that the -Br ought to be more labile, so perhaps refluxing with N-chlorosuccinimide might
replace the -Br with -Cl and trap the bromine as NBS.
Simply bubbling in Cl2 sets up an equilibrium with ClBr and CCl4 if I recall, and bubbling in HCl similarly makes an equilibrium with chloroform and
Br2. Anyway neither is very satisfactory preperatively.
Photochemical chlorination with Cl2 needs special filtering of the UV to preclude side reaction forming hexachloroethane. Kopp Glass makes such
filters, not cheap!
I am tempted to try the wet iron filings routine, this gets chloroform from CCl4 - I am hoping it would selectively reduce the -Br leaving chloroform
and not CHCl2Br.
Any ideas anyone?
[Edited on 16-10-2008 by Sauron]
Sic gorgeamus a los subjectatus nunc.
|
|
|
Slimz
Hazard to Others
Posts: 123
Registered: 18-9-2007
Member Is Offline
Mood: Inquisitive
|
|
Chloroform is way easier to make than all that..
NaClO + dimethyl ketone (Acetone) you get a layer of chloroform.
thats 500ml of bleach (normal strength) and 5ml of acetone.
a stronger solution of bleaching powder and water can be used but you should keep the reaction cooled in a bin of ice if doing that
Johnny was a chemist’s son, but now he is no more. What Johnny thought was H2O was H2SO4
|
|
|
Sauron
International Hazard
Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
Go teach your grandmother to suck eggs, slimer.
I do not want to muck around with haloform reaction.
Back to the topic:
The Ph3P reaction with CCl4 or CBr4 to halogenate alcohols or carboxylic acids, ought to work with CCl3Br ought it not? I wonder if it might leave
CHCl3 abd an alkyl bromide (or acyl bromide) in this case?
Also, the recently discussed NaBH4 reduction of polyhalomethane, I wonder whether it might be selective in a case like this? Would the product be
CHCl3 or CHCl2Br? or a mixture of both?
[Edited on 16-10-2008 by Sauron]
Sic gorgeamus a los subjectatus nunc.
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Not too familiar with such compounds, maybe zinc dust would cause selective dehalogenation?
How could you differentiate CCl3Br from CCl4? IR? Or is there a major difference in bp?
Maybe CClBr is interchangeable with CCl4 is certain applications (what do you plan on using the tetrachloride for?), and if it is much more reactive,
this is surely du to the Br, so you could play on that to selectively remove/exchange it...
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
Sauron
International Hazard
Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
Significant difference in MW, BP, density, refractive index. Give me a moment to collect the data and I will post it here.
Synonyms: Tetrachloromethane
Molecular Formula: CCl4
Formula Weight: 153.82
Registry number: 56-23-5
MDL number: MFCD00000785
Density: 1.594
Melting point: -23 °C (lit.)
Boiling point: 76 °C (lit.)
nD20: 1.459-1.461
Synonyms: -
Molecular Formula: CBrCl3
Formula Weight: 198.27
Registry number: 75-62-7
MDL number: MFCD00000783
Density: 1.997
Melting point: -6 °C (lit.)
Boiling point: 105 °C (lit.)
nD20: 1.5055-1.5075
See what I mean?
No troule to fractionate. Can probably fractionally crystallize as well.
[Edited on 16-10-2008 by Sauron]
Sic gorgeamus a los subjectatus nunc.
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Indeed, quite convenient. i was rather thinking of following the reactiosn though that direct workup, to determine the best tempa nd reaction times
etc. I guess the refractive index can come in handy if you have the necessary equipement.
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
Sauron
International Hazard
Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
An Abbe refractometer is on my list of things to buy.
But seems to me that given a spread from 1.59 to 1.99 in density, that a set of hydrometers (which I have) will also quickly and easily determine any
mixture quantitatively to decent accuracy.The hydrometers read to 3 or 4 decimel places.
The way that CCl3Br is made is to treat CCl4 with AlBr3. Maybe treating the mixed tetrahalide with AlCl3 might reverse that?
Any mechanism that proceeds via the trichloromethyl radical needs to be shunned or hexachloroethane will result.
Sic gorgeamus a los subjectatus nunc.
|
|
|
Sauron
International Hazard
Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
The Ph3P reaction appears not to be selective, or at best partially selective. The famous "One-Pot Conversion of Alcohols to Amines" which is on all
the notorious druggie sites has the system Ph3P-CBrCl3 producing mixed alkyl chlorides and bromides. They prefer the bromides for higher reactivity
with NaN3.
I have found a totally selective reducing agent that takes bromotrichloromethane to chloroform. But it remains to be seen if this has any practical
preperative utility.
2,6-dimethyl-3,5-dicarbomethoxy-1,4-dihydropyridine, a derivative of 2,6-lutidine, is the reducing agent, and is oxidized to the corresponding
pyridinium bromide. This is a free radical reaction and was first observed with CCl4 while trying to obtain NMR spectra. The reaction is slow, but
later that with bromotrichloromethane was found to proceed more rapidly.
The prep of this compound is in a 1914 paper in Monatsh. by Hans Meyer and Hans Tropsch. I have requested it. So more to follow later.
[Edited on 17-10-2008 by Sauron]

Sic gorgeamus a los subjectatus nunc.
|
|
|
Sauron
International Hazard
Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
The compound shown above is also the parent structure for the potent coronary vasodilator and hypotensive agent nifedipine, which differes from it
only by having a 2'-nitrophenyl group at the 4-position.
OK now I have Bayer's patent, the 1914 Monatsh.article, and at first glance this looks fine. Condensation of two mols acetoacetic ester with 1 mol
formaldehyde and 1 mold ammonia. However the devil is always in the details so let me digest these before commenting further. I also found a nice
Chem.Rev piece on the 1,4-dihydropyridines.
I will post these after perusing.
By the way the condensation of acetoacetic ester (2 mols), an aldehyde and an amine to form 1,4-dihydropyridines is the Hantzsch reaction and these
compounds are often reffered to as Hantzsch-type structures,
[Edited on 17-10-2008 by Sauron]
Attachment: cr60275a001.pdf (946kB)
This file has been downloaded 3048 times
Sic gorgeamus a los subjectatus nunc.
|
|
|
Sauron
International Hazard
Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
Actually the Hantzsch Dihydropyridine Synthesis is better stated to be the condensation of an active methylene compound (two mols) with ammonia or a
primary amine, and an aldehyde.
There's also the Knoevenagel-Fries modification which allows for prep of unsymmetrical dihydropyridines.
But this target of ours is simple classical methyl acetoacetate, formaldehyde, and ammonia. I suspect paraformaldehyde etc works.
Hexamethylenetetramine has been used. The reaction is sone in methanol and yields are good to excellent.
In the case of that anti-angina drug, 2-nitrobenzaldehyde replaces the formaldehyde. Simple as that. The Bayer US patent is attached, the examples
give a good idea of the ease of preparation of this class of compounds.
When used to reduce CCl3Br to chloroform, the dihydropyridine is oxidized to the aromatic pyridinium bromide quaternary salt. Fortunately this can be
reduced and reused. Whether it is easier to just make more, remains to be seen. The usual reducing agent is sodium dithionite.
Org Syn website was down for a couple days but is now up again. A Hantzsch synthesis of shown for 2,6-lutidine (2,6-dimethylpyridine) in which the
initial product is the ethyl ester form of our target. (2,6-dimethyl-3,5-dicarbomethoxy-1,4-dihydropyridine.) Their prep employs ethyl acetoacetate
and aqueous formaldehyde soln (formaline) and anhydrous ammonia.
They then oxidize and decarboxylate the Hantzsch compound to obtain the dimethylpyridine.
Looks good to me.
[Edited on 18-10-2008 by Sauron]
Attachment: US3485847.pdf (243kB)
This file has been downloaded 743 times
Sic gorgeamus a los subjectatus nunc.
|
|
|
Sauron
International Hazard
Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
Here is the Org Syn prep of 2,6-lutidine, or dimethylpyridine. This is a pretty typical Hantzsch reaction although the authors did not even tip their
hats to his pre 1890 work.
I am not so sanguine about the 10 drops of Et2NH, or the delay before introducing NH3. There are alternatives to admitting ammonia as anhydrous gas,
notably ammonium acetate.
You can ignore the second half of the prep, unless you want to use the Hantzsch as they did to prepare pyridines instead of dihydropyridines.
[Edited on 19-10-2008 by Sauron]
Attachment: CV2P0214.pdf (135kB)
This file has been downloaded 705 times
Sic gorgeamus a los subjectatus nunc.
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Are you sure NH3 can be replaced by ammonium acetate here? I guess it simialr to some kind of mannich reaction, but you might want to check if there
is a simialr procedure using an ammonium salt in the particular variant.
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
Sauron
International Hazard
Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
Go a few posts up and download the Chem Rev monograph.
There's more than one way to add formaldehyde, and more than one way to add ammonia.
And alternatives to acetoacetic ester(s) albeit not for this particular product.
The Chem Rev paper also references other reviews. There's a Wiki page on the Hantzsch reaction, and it has some refs as well.
The Hantzsch is like the Mannich reaction in having three components, but is a lot more facile than the Mannich which requires very fiddly control of
pH. There's absolutely no indication of such requirement here. It seems to be, chill mix, stir, let sit for 2 days. seperate, dilute, chill
again,saturate with NH3, let sit again for 2 days. Filter and dry. The hardest thing about this prep is waiting 48 hrs x 2.
While I can buy anhydrous NH3, the SS regulator is expensive, hence my interest in an alternative.
Also see the patent I posted which replaces formaldehyde with benzaldehyde to prepare nifedipine, it may contain some variants on how to get the N in
the ring as well.
One of the Pyridine volumes in the Chemistry of Heterocyclic Compounds series (which most of are now available for download from References) covers
this material although it is organized from the pyridine not dihydropyridine standpoint.
I believe ammonium acetate is introduced along with NaOH and forms NH3 in situ. NaOAc salts out.
[Edited on 19-10-2008 by Sauron]
Sic gorgeamus a los subjectatus nunc.
|
|
|
Sauron
International Hazard
Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
Here is the original, 1881, two page short and sweet paper on what became known as the Hantzsch Pyridine (Dihydropyridine) Synthesis.
Ber., 14, 1637-1638 (1881) cortesy of FREE BnF Gallica, and nuts to Wiley!
[Edited on 19-10-2008 by Sauron]
Attachment: Hantzsch1881.pdf (109kB)
This file has been downloaded 812 times
Sic gorgeamus a los subjectatus nunc.
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Ok, perfect then. I was thinking you were planning on using the ammonium salt directly to the acetoacetic ester. This really does look like a evry
handy reaction, and I'm wondering what other uses we could find for the dehydropyridine, as a mild and selective reducing agent.. The fact that it can
be recyle easily seems very promising, and perhasp it could be used in catalytic amounts, adding dithionite gradually to the compound to be
reduced/dehalogenated and the dehydropyridine, reducing the pyridine bromide as it is formed..
EDIT: I've just realized I've read about this reaction last night in the "Chemistry of Enamines", from the FTP, jump to page 543 (on the DejaVu menu),
and you will see that enaminones can be used in the reaction, so the ammonia can be added tot he acetoacetic ester before the formaldehdye.
Apparently, heating the enaminone with benzaldehdye, benzoic acid and ammonium acetate gives a tetrahydropyridium acetate, which could be either
reduced and decarboxylated to a 4-amino piperidine, or maybe dehydrogenated to the pyridine, and decarboxylated (route to DMAP?):
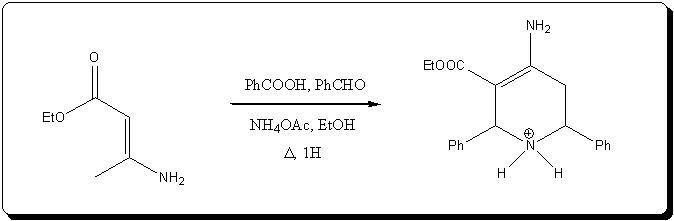
Ref to the enaminone variant:
- F. Micheel, H. Dralle, Justus Liebig's Ann. Chem.; 670, 57 (1963)
-A. H. Cook, I. M. Heilbron, L. Steger, J. Chem. Soc.; 413 (1943)
-U. Kuckländer, P. Ulmer, G. Zerta, Arch. Pharm. (Weinheim); 322, 437 (1989)
[Edited on 19-10-2008 by Klute]
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
Sauron
International Hazard
Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
It gets better. Apparently the reduction is a free radical one and only a tiny amount of the dihydropyridine is required to reduce a much larger
amount of the CBrCl3.
The reaction can be initiated photochemically or thermally and while they were doin this in methanol for kinetics studies, the paper states that the
reaction also works in nest bromotrichloromethane, but the ppt of pyridinium bromide made rate measurements difficult. Well, we don't care about
kinetics so methanol be damned.
I'd better post the paper I am paraphrasing so you can suss it out yourself. You just ignore all the deuterated stuff and the pchem malarky. Bottom
line is that this is not a stoichiometric reaction and not much dihydropyridine is needed at all. Fine with me.
[Edited on 19-10-2008 by Sauron]
Attachment: cbrcl3red.pdf (695kB)
This file has been downloaded 788 times
Sic gorgeamus a los subjectatus nunc.
|
|
|
jarynth
Hazard to Self
Posts: 76
Registered: 12-8-2008
Member Is Offline
Mood: No Mood
|
|
| Quote: | Originally posted by Sauron
Here is the Org Syn prep of 2,6-lutidine, or dimethylpyridine. |
| Quote: | | You can ignore the second half of the prep, unless you want to use the Hantzsch as they did to prepare pyridines instead of dihydropyridines.
|
From the last step it appears that the decarboxylation of pyridinecarboxylic acids can be effected in good yields without the use of copper chromite
catalysts. Klute, this might be something for your pyridine from niacin experiments.
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Sorry to say this Jarynth, I'm not the one that was trying to prepare pyridine by decarboxylation  I beleive it was Ordenblitz. I beleive it was Ordenblitz.
But I have been pretty interested in preparing dimethylaminopyridine from cheap starting materials, although for the moment it's been nothing but a
paper distraction..
In any case, forming a piperidine ring from acetoacetic ester seems like a very good option. Possibly benzaldheyde could be replaced by formaldehyde
in the recation I described above.
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
Sauron
International Hazard
Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
In one of the papers I have, 2,6-dimethylpyridine-3,5-dicarboxylic acid is oxidized to 2,3,5,6-pyridinetetracarboxylic acid, and the 2 and 6 positions
decarboxylated. The product is dinicotinic acid. I think you will find that the 3 and 5 positions, beta to the ring N are very hard to decarboxylate.
Maybe better to degrade nicotinamide to 3-aminopyridine and get rid of the amino group via diazotization, is what you want is pyridine. Seems like a
lot of work just to make pyridine.
You might consider making citrazinic acid from citric acid, haloginating the 2,6-hydroxyls and then getting rid of the halogen by reduction. Voila,
pyridine from citric acid. See my thread on citrazinic acid from last year.
Well, nicotinamide (niacin) is cheap, I have a Kg of Roche pharm grade sitting here.
Sic gorgeamus a los subjectatus nunc.
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
No, no, no! I don't want to make pyridine I just buy the stuff. I was more
interested in things like 4-aminopyridine, although the nitration of pyridine-N-oxide and subsequent reduction look much more pratical after all. It
would ahev been nice to have a reaction starting from something else than pyridine as the stuff reeks..
The 3 and 5 position are decarboxylated with Ca(OH)2 when preparing 2,6-dimethylpyridine, in the OrgSyn pre you posted. Are you not confusing with
the 2 and 6 carboxyl?
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
panziandi
Hazard to Others
Posts: 490
Registered: 3-10-2006
Location: UK
Member Is Offline
Mood: Bored
|
|
I was looking into making fluorine compounds myself and found that reaction of tri-iodomethane with silver fluoride (can also be mercury fluoride and
several others too) gives various fluoro-iodomethanes along with trifluoromethane sometimes explosively! Now further research revealed the use of
antimony trifluoride. Exchanges of I and Br for F worked. Perhaps Cl3C-Br could be converted to CCl4 by the use of silver chloride or antimony
trichloride. Now I don't know if this would work, there wouldn't be much literature (I haven't found any) on this proposed reaction mainly because
CCl4 and CHCl3 are produced by far more economical routes industrially. But perhaps Sauron, if you could spare a little Cl3C-Br for experimenting, you
could try reacting it with silver chloride, mercury chloride or perhaps antimony trichloride? Perhaps it would require reflux or perhaps heating in an
autoclave?
|
|
|
Sauron
International Hazard
Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
CBrCl3. Anyway I have a folder on fluoform somewhere. If memory serves isn't it a gas?
Klute, I just buy pyridine too.
Are you sure you want to prepare DMAP? It is a very very toxic Ca-channel blocker, the LD50 is quite appalingly low. That's why they sell it in
prills, to make handling safer.
Sic gorgeamus a los subjectatus nunc.
|
|
|
panziandi
Hazard to Others
Posts: 490
Registered: 3-10-2006
Location: UK
Member Is Offline
Mood: Bored
|
|
Cl3C-Br makes it clearer to some of the people potentially reading this that what bond im talking about.
Indeed many of the fluoromethanes are gases. The reactions are done in steel setups and the products cool in dry ice traps I had diagrams in one paper
I found. But I don't see any real reason why an analogous reaction with chlorine compounds couldn't work. The driving force is the lattice energy of
the AgI vs AgF I believe. Antimony trifluoride was used to replace the first two fluorines as using heavy metal fluorides was too vigorous, then
silver or mercury fluoride was used to replace the last iodine. If you used silver chloride and fractionated/refluxed at the same time you could
remove the CCl4 as it was formed. Perhaps it is worth a small scale experiment to see if it is feasible?
|
|
|
Sauron
International Hazard
Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
Antmony trichloride difluoride is often used in lieu of the uoride or the pentafluoride. See Brauer for preparation.
triflAgF and HgF are very old (19th century) reagents. CoF3 is more contemporary and the above mentioned SB compounds intermediate in vogue.
You can write it as you see fit but I desribed the substance as bromotrichloromethane throughout so I do not think alongside that, CCl3Br or CBrCl3
are at all ambiguous. C-C-I3-Br makes no sense at all. So a tetrahalomethane is all it can be, especially when reduced to chloroform, or chlorinated
to CCl4.
[Edited on 19-10-2008 by Sauron]
Sic gorgeamus a los subjectatus nunc.
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Well, Panreac and Acros sell DMAP in small format (20-25g) at a reasonable price, but I was more interested in trying to prepare a polymer-supported
version (like I love to do with any catalyst I hear about), the commercial poly-DMAP is incredibly expensive. A supported version would have several
advantages, like being very easy to seperate and re-use, as most heterogenous catalysts, but also making it must less easily absorbed by skin and thus
much easier to handle and supposebly less toxic. But I'm already scattering myself over several projects, so I'll keep this for a distant period.
Sorry if I'm diverting the topic, I'll stop there.
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
| Pages:
1
2 |










