Synthetic routes to tetrazole compounds based on OTC materials.
As one may know tetrazoles are class of highly energetic nitrogen based materials. This compounds combine an unique
set of properties, attracting much attention in modern research for primary explosives. However relatively difficult preparation of source products,
made this extremely interesting class of compounds hardly accessible for amateur chemist. However it will be shown below that even such complex
compounds can be made in home with little effort. The main source product for all variety of tetrazoles is 5-aminotetrazole, witch can be made in two
general ways – by Schtolle’s method (from cyanamide or dicyandiamide and hydrogen azide) and by Thiele’s method (by diazotizing aminoguanidine
with nitrous acid in strong mineral acid medium). Since sodium azide is not common in amateur chemistry due to high cost and toxicity, route through
aminoguanidine seems much more laborious for amateur chemist use. Genetic structure of 5-aminotetrazole is shown on scheme below:
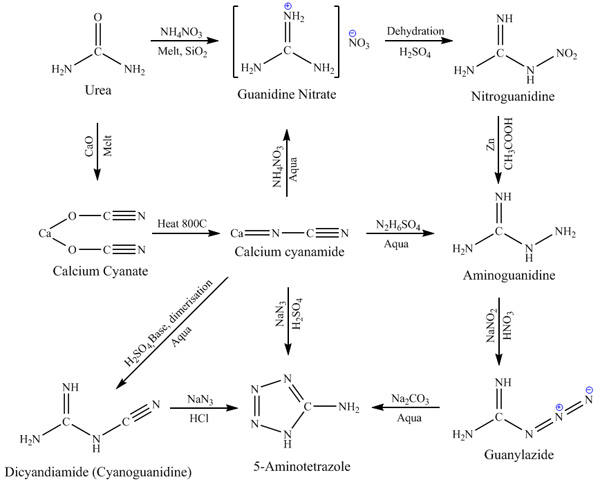
Aminoguanidine itself can be produced in two different routes, both including intermediates witch can be interesting
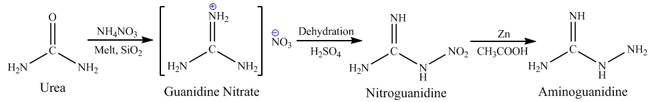
not only in terms of production of aminoguanidine but also by themselves. First route is nitroguanidine route, it consists in melting urea with
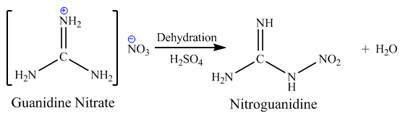
ammonium nitrate in presence of silicagel catalyst to produce guanidine nitrate, dehydration of guanidine nitrate by sulfuric acid to produce
nitroguanidine and reduction of later by means of zinc dust in acetic acid. Main disadvantages of this route are: explosion hazard of melting urea and
ammonium nitrate if reaction conditions are not controlled property, quite difficult nitroguanidine reduction procedure requiring zinc dust and
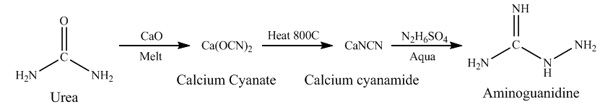
glacial acetic acid. Second route is cyanamide based, it consist in melting urea with calcium oxide to produce calcium cyanate witch can be decomposed
at high temperature to form calcium cyanamide, reaction of later with hydrazine sulfate in aqueous media leads to formation of aminoguanidine. The
main difficulty on this route is requirement of high temperature and some additional efforts to isolate cyanamide from the air oxygen in course of
production to produce high quality calcium cyanamide required by final step. Below i will describe both reaction routes as well as procedure of
conversion of aminoguanidine to 5-aminotetrazole in final section. I hope this materials will help reader to find his personal route to produce and
study this interesting classes of nitrogen organic compounds.

Chapter 1 – Aminoguanidine, cyanamide route
Step №1. Constructing 900C electric furnace. In general the oven consists of ceramic
fireproof muffle tube, covered by nichrome wire heating element, covered by radial fireclay layer which ensures that main heat flow is directed to
opposite direction – into the muffle. To prevent heat losses to environment and allow heating to high temperature the whole construction is
protected by heat shielding. Temperature in the muffle is measured by means of thermocouple. Heat wire is powered by AC power supply which allows
voltage regulation – this will allow temperature regulation inside the muffle by regulating heat flow from heat element.
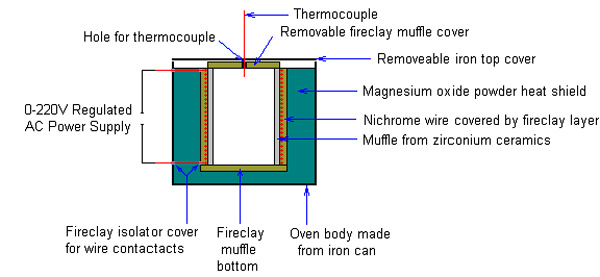
Heating Element. Heating element consists form fireproof ceramic muffle, covered with nichrome
wire of long lenth. Materials for furnace muffle must be stable at high temperature, have high electric resistance and resistance to heat shock.
Ceramic tubes from ZrO2/MgO/Al2O3 based ceramics are essential for the job. In will be impossible or very hard to make this one by yourself, so search
for ready ones for example in high voltage safety fuses and other high voltage components containing ceramic parts. Purpose of wire is to produce much
heat then current passes through so it must have high electric resistance, weakly dependent from temperature (this will allow better regulation of
heat flow by changing current) and be highly corrosion resistant to air oxygen at high temperature. Usual material witch satisfies this requirements
is nichrome (80:20 nickel/chromium alloy), allowed working temperature of such alloy is dependent from wire diameter and is approximately 1000C.
In my furnace i’ve used muffle tube made from zirconium oxide bases ceramics, with 9 cm lenth, 6 cm outer diameter
and ~4 mm wall thickness. This ceramic muffle is wound with narrow equally space turns of nichrome wire with 0.5 mm diameter. In order to make turns
equally spaced, the wire is wound in pair with filament having approximately the same diameter, making this double turns contiguous to each other,
after all turns have been wound, filament is carefully removed leaving fine coil with equally spaced turns. Remember to leave some free edges to wire,
those will be needed to connect wire to power supply. My coil with 81 Ohm electric resistance is shown in photo section below.
Calculation of power parameters. Now then we have heating element, we can measure resulted wire
resistance and use it to calculate working power parameters. This moment is very important since if we allow too strong current to the wire it will
burn out, if current will be too low we will not get enough heat to reach high temperature. Heat release in wire is described by Joule–Lenz,
according to witch heat flow is proportional to square of current multiplied by wire resistance:
P = (I^2)*R = (U^2)/R
Wire of each diameter has certain maximum allowed current and recommended work temperature, both are higher for wire
of higher diameter, however it should be remembered that wire of relatively high diameter will also require much more current to produce good heat
flow – this will lead to complications with power supply. It’s a good idea to use wire of some optimum diameter, for general heating devices this
diameter is about 0.4-0.6 mm. Table below shows maximum allowed current and recommended work temperatures for wires of different diameters:

My wire was 0.5 mm diameter, and according to table has maximum allowed current of 3.5A and recommended work
temperature about 1000C. If current passing the wire will be higher then allowed wire will burn out, while values close to this value will lead to
decrease of wire durability, so this is a good idea to use current slightly below allowed maximum – for example 0.8 of maximum allowed current, for
my wire this limit current will be 3.5*0.8 = 2.8A. Now then we determined maximum work current we can use this value along with wire resistance to
calculate maximum work voltage, and substitute resulted value to Joule–Lenz equation to calculate maximum heat power:
Umax = Imax * Rwire = 2.8 * 81 = 226.8 V
Pmax = (Umax^2)/Rwire = (226.8^2)/81 = 0.635 KW
Muffle coating. Now then our heating element is ready we must provide if with coating. Coating is
essential not only to fix wire in place, but also to insure most part of heat flow will be directed to inside of muffle. To do this we must coat our
heating element with 0.5-1cm layer of fireclay, I’ve used layer with approximately ~0.7 mm thick. Take some fire clay and add the least water needed
to reach plasticity and cover wire with a clay layer, then the wire is covered and the clay layer is smoothed by rolling on a smooth surface. The
whole device is placed into a kitchen oven to dry for 2 hours at ~250C. When initial drying is finished, attach contacts of nichrome wire to power
supply and heat it slowly rising voltage to nominal and allow element to sit at this voltage for a while (0.5-1 hour) to complete dry clay layer. Same
clay mass is used to make top and bottom caps for the muffle, however this does not require so careful tempering and can be used after 1-2 hours of
drying in kitchen oven.
Furnace body. To make whole element more comfortable to work and make it more durable it is placed
to the iron can of appropriate diameter, witch is supplied by two drilled holes for wire outlets. Remember to electrically isolate such holes, because
if this not done it will lead to short circuit through the iron can body. Bottom of the can is filled with moderately thin layer of magnesium oxide,
clay bottom cap is placed to the center. Heating element is placed axially to the can body on the top of the bottom cap and whole space between walls
of can and heating element coating is filled with powdered magnesium oxide. This layer of magnesium oxide forms primary heat shield for the furnace,
while iron can ensures integrity of furnace in case of coating cracks (this is usually the case if furnace completed several heating/cooling cycles,
and appears to be unavoidable while using commonly available materials for muffle coating). Don’t forget to supply furnace with top clay and iron
can caps, to prevent heat loss through the top of the furnace, those can be provided with hole drilled in the center – for thermocouple temperature
control. For my furnace i’ve use iron can from conservated food with 10 cm diameter and height of 12 cm.
Additional Heat Shielding. Your furnace is ready to work, however to reach high temperature heat
shielding should be extended. This can be achieved by placing element to the center of wider saucepan and fill free space between with wide layer of
powdered magnesium oxide. My furnace can was ~10 cm diameter and i’ve used saucepan with 16 cm width. Now then entire device is assembled, you can
provide it with thermocouple and connect it to the suitable AC power supply. I’ve used a laboratory auto transformer (0-220V AC), with maximum
secondary coil current of 4A. Assembled device of type I’ve described was able to reach temperature of 900C in less then 1.5 hour.
Photos:
Step №2. Preparation of calcium cyanamide from urea and calcium oxide.
Properties. Pure calcium cyanamide is a colorless crystalline solid, soluble in water (2.5g in 100
ml of water at 25C) and insoluble in alcohol. It is volatile at high temperatures, sublimes at 1090C (at atmospheric pressure), and can be melted
under excessive pressure of nitrogen at 1340C. Dissolved in cold water, it is hydrolyzed forming a soluble acidic salt and calcium hydroxide (eq.1),
while hot water decomposes calcium cyanamide with formation of urea and calcium hydroxide (eq.2), and action of water steam at 110-115C causes release
of ammonia and formation of calcium carbonate (eq.3). At temperatures above 400C calcium cyanamide is readily oxidized by air (eq. 4).
2CaCN2 + 2H2O(cold) → Ca(CN2H)2 + Ca(OH)2
CaCN2 + 3H2O(hot) → Ca(OH)2 + CO(NH2)2
CaCN2 + 3H2O(steam) → CaCO3 + 2NH3
2CaCN2 + 3O2 → CaCO3 + 2N2
Pure calcium cyanamide contains 34.98% of nitrogen. The industrial product has greyish black color due to
contamination by carbon (9-13%), contains 55-65% of pure cyanamide and 18-24% of nitrogen. Additional contaminants (~5%) contain mainly silicic acid,
oxides of iron, magnesium and aluminum. In addition industrial calcium cyanamide contains 14-20% of calcium oxide and 2-5% of unreacted calcium
carbide. Calcium cyanamide slowly reacts with moisture in the air forming urea, cyanamide, dicyandiamide and ammonia. Calcium cyanamide is used in
agriculture as a nitrogen fertilizer. In chemical industry calcium cyanamide was used to produce cyanides, guanidine and a large variety of organic
compounds. Calcium cyanamide is toxic and acts mainly as a skin and lung irritant. Toxicity is relatively low but can be raised greatly then combined
with alcohol. Action of cyanamide dust followed by alcohol drinks cause severe poisoning accompanied by agonising suffocation. This specific toxic
effect is reason why cyanamide solutions are proposed for use in anti alcoholism drugs.
Preparation. Calcium cyanamide is produced on an industrial scale by fixation of atmospheric
nitrogen by calcium carbide at temperatures above 1000C in electric furnaces:
CaC2 + N2 → CaCN2 + C
Other methods of preparation include: action of ammonia on cyanogen chloride, melting of urea with metallic sodium,
high temperature reaction of hydrocyanic acid with calcium oxide and thermal decomposition of calcium cyanate. It is clear that only last one is
possible in home scale since calcium cyanurate can be readily produced by melting urea with calcium oxide or hydroxide, but this method require some
work to gain access to high temperature. The process consists of two main stages – in the first stage is formation of calcium cyanate by melting
urea with calcium oxide. Thermal decomposition of urea is not a simple process and can produce wide variety of products, depending from reaction
conditions and timing, but in presence of alkali it goes almost exclusively to cyanate direction. Mechanism of cyanate formation can be expressed by
the following reactions:
1. 3CO(NH2)2 → 3HOCN + NH3
2. CaO + 2HOCN → Ca(OCN)2 + H2O
3. HOCN + H2O → CO2 + NH3
As one can see, theory requires 3 moles of urea for 1 mole of calcium oxide, since one molecule of cyanic acid is
decomposed by water liberated in neutralization of calcium oxide. Reaction takes place at temperatures above urea melting point (120 - 350C) and
causes formation lots of ammonia gas, accompanied by intensive foaming of the melted mixture. When the process is nearing completion the reaction
mixture re-solidifies due to formation of calcium cyanate which have higher melting point (decompose without melting at higher temperatures). In order
to produce calcium cyanamide, calcium cyanate produced is cooled, grounded in mortar and calcined at high temperature in absence of air oxygen. The
second stage (calcination of calcium cyanate) proceeds at temperatures about 700-900C, producing calcium cyanamide and carbon dioxide (Eq.1 below). It
should be noted that that air oxygen oxidizes produced cyanamide to carbonate (Eq.2 below), and process should be carried out with very limited air
contact or under protective gas atmosphere:
1. Ca(OCN)2 → CaNCN + CO2
2. 2CaCN2 + 3O2 → CaCO3 + 2N2
Procedure. Preparation of calcium cyanamide was performed according to US patent 5753199. Prepare a
mixture of 56.4g (1 mol) of pure finely ground calcium oxide (Note №1) and 180g (3 mol) of pure fine urea. Mixture is placed to saucepan and
heated on a hotplate. As soon as the urea melts the reaction starts, a lot of ammonia is evolved and mixture begin to foam (Note №2). After
foaming subsides, mixture begins to solidify and became unstirable. Then reaction mixture is completely solidified, heat source is turned off and
still hot product is grounded to fine powder. This yields about 134g of crude calcium cyanate (Note №3). Calcium cyanate is then placed to
ceramic crucible, witch is covered by closely fit clay lid, placed to electric furnace (also provided with closely fit cover) and is heated up to
750-800C for one hour (Note №4). After this period furnace is turned off and is allowed to cool down, without opening furnace cover. Then
temperature in furnace is downed to 150-200C, furnace cover and crucible is removed and placed to cool on the air. Opened crucible is filled with
spongy pinkish-white calcium cyanamide, with small (about ¼ of crucible) bottom layer of product with grey color (Note №5). Total yield is
69.3g of high quality calcium cyanamide (86% from theory), containing almost theoretical amount of nitrogen (~33-34%).
Notes:
1. It is essential to use pure calcium oxide, to prevent presence of alkali metal salts in reaction
mixture. Those at first will form corresponding cyanates, witch are unstable at high temperature of second – calcinations stage and will decompose
to form cyanide and oxygen. Even if relatively small amounts of sodium or potassium are present, final product can became much more dangerous since on
action of acids it will realize extremely dangerous and toxic hydrogen cyanide, in quantities easily detectable by smell from such reacting mixture,
or even from solid product itself.
2. Foaming of reaction mixture is very intensive and can easily overflow reactor wessel. This must
be taken to account then choosing volume of reactor. Foam can be sufficiently settled down by intensive mixing. Note that moistured source materials
will not result in even more intensive foaming but will also reduce overall yield due to increased moisture decomposition of cyanic acid.
3. This quantity is larger then theoretical (134g vs 124g theory), this is result of the fact that
solidification and drop of reaction rate occur earlier that source products are completely reacted. It was found that continue of heating after
solidification doesn’t lead to sufficient change of mass and complete transformation. Analysis of reaction product shown clear strong signal for
cyanate, but some small amount of unreacted source products also present. This however is not critical for further calcinations step, since strong
heat will cause them to react further, forming final reaction product.
4. Then this crude cyanate is heated up to about 300-400C, impurities of unreacted urea and calcium
oxide will react/decompose, forming more ammonia. This additional amount of ammonia evolved is quite small but is easily detectable by smell, so it is
advised to perform calcinations in place with good ventilation. Remember to use closely fit lids for both crucible and furnace to prevent oxidation by
air, if such lids are used circulation of waste reaction gasses is avoided, allowing them to act as protective atmosphere for main product.
5. Analysis of both colored layers shown that their constituents are exactly the same – both show
strong and clear signal for cyanamide with ammoniacal silver nitrate and both contain only tiny traces of cyanate shown by reaction with cobalt salt
solution. It appears that grey coloration of bottom layer is caused by charring of some organic impurities present in starting cyanate, and is not
related to exact calcination temperature or speed of heating.
Photos:
1. Photos show reaction mixture on beginning of foaming and at solidification point, grounded
reaction product and it's reaction on cyanate using cobalt salts.
2. Photos below shows two layers in product received after calcination, reaction of both layer
samples on cyanamide using ammoniacal silver nitrate and final product with and without flash.
Step №3. Preparation of aminoguanidine bicarbonate from calcium cyanamide and hydrazine sulphate.
Properties. Aminoguanidine is crystalline compound unstable in free state, perfectly soluble in
water and insoluble in alcohol, melting point not determined because heat causes decomposition before melting point. Aminoguanidine shows basic
properties and forms stable salts with strong acids via amino group, many from them form yellowish solutions in water. Free aminoguanidine and
alkaline solutions of it’s salts are unstable to heat and oxidation by air oxygen, oxidized products provide such solutions with intense red
coloration.
Derivatives. Aminoguanidine hydrochloride, NH2C(=NH)NHNH2*HCl, can be obtained by gradually adding
glacial acetic acid (124 grams) diluted with an equal volume of water, to a mixture of nitroguanidine (208 grams) and zinc-dust (700 grams) which has
been previously rubbed to a thick paste with ice and water; during the addition of the acid, which occupies 2-3 minutes, the mixture is constantly
stirred, and the temperature kept at 0°C by adding ice. The temperature of the mixture is then allowed to rise slowly to 40-45°C, and, as soon as a
portion givers no coloration with soda and a ferrous salt, reduction is at an end; the filtered solution is then mixed with excess of hydrochloric
acid, concentrated on the water-bath, taken up with alcohol, and evaporated to dryness; the residue consists of aminoguanidine hydrochloride,
guanidine hydrochloride, and a little ammonium chloride, which are separated by means of alcohol. Aminoguanidine hydrochloride crystallizes from
dilute alcohol in large, thick prisms, melts at 163°, and is very readily soluble in water, but insoluble in ether. The platinochloride,
(CH6N4)2*H2PtCl6, is a yellow substance melting at 145 - 146°C.
The nitrate, NH2C(=NH)NHNH2*HNO3, crystallizes from water in large plates, from alcohol in needles, and melts at
144°C; it is only sparingly soluble (12 parts in 100) in water at 16°C. The sulphate, (CH6N4)2*H2SO4*4H2O, crystallizes in needles, loses its water
at 110°C, and melts at 207-208°C with decomposition ; it is very readily soluble in water, but insoluble in alcohol. The acid sulphate, CH6N4*H2SO4,
crystallizes from water, in which it is only sparingly soluble, in small, yellow needles. When a solution of the sulphate is treated with the
theoretical quantity of barium hydroxide, a solution of aminoguanidine is obtained; this solution gradually turns reddish on exposure to the air, and
decomposes, with evolution of ammonia, when evaporated at a moderate temperature; on evaporation under reduced pressure, it yields a reddish,
crystalline substance which has an alkaline reaction, and is soluble in alcohol, but insoluble in ether. A compound of the composition
(CH5N4)2Cu*2HNO3, is obtained as a violet, crystalline precipitate when a solution of aminoguanidine nitrate is treated with copper nitrate and sodium
acetate; it crystallizes in microscopic plates or prisms, and is only very sparingly soluble in cold water, yielding a violet solution; it is
decomposed by boiling water with separation of copper, and also by ammonia and nitric acid. The corresponding sulphate, (CH5N4)2Cu*H2SO4, prepared in
like manner, is a violet, crystalline, sparingly soluble powder.
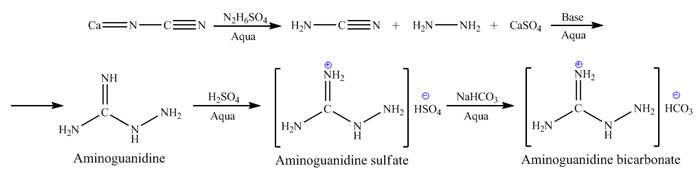
Preparation: Preparation of aminoguanidine was carried out on base of procedure described in US
Patent 3673253, and is based on condensation of cyano-group in free cyanamide with hydrazine. Reaction of hydrazine sulphate with calcium cyanamide in
aqueous media results in formation of insoluble calcium sulphate and liberation of free cyanamide and hydrazine, witch react with each other in basic
media forming aminoguanidine. Since cyanamide is quite reactive and can readily form dimeric form – dicyandiamide witch doesn’t produce
aminoguanidine on reaction with hydrazine, this side reaction leads to decreased yield of final product. Reaction conditions, mixture pH and
temperature are controlled in such manner witch allows to overcome this side process. This is achieved by keeping minimal concentration of cyanamide
and decreasing pH by addition of sulpuric acid to level keeping conversion to dicyandiamide at minimum while still allowing fast condensation process
for cyanamide and hydrazine. Reaction scheme is shown on picture below:
Procedure:55.8g of monohydrazine sulphate (0.429 mol) and 40g of finely ground calcium cyanamide
containing 30% N (~0.5 mol) were simultaneously and uniformly added with vigorous stirring to 200 ml of water at pH value of 9.5 (Note №1). The
pH value was regulated by the addition of 50 percent sulfuric acid in an appropriate quantity (Note №2). The reaction temperature was kept at
40C. After reaction mixture had been stirred for 30 minutes at pH 9, the pH value was adjusted to 7 and the mixture was heated for about 1 hour at 80C
at that pH value. After reaction mixture had been cooled to 60C, it was filtered and the filtration residue, consisting essentially of calcium
sulfate, was washed with 200 ml of warm water (Note №3). Both the filtrate and water used for washing were collected in a precipitation vessel
and were adjusted to a pH value of 6.5 with 50 percent sulfuric acid. 36.9g of sodium bicarbonate were then slowly and uniformly added at 30C as a
result of which the aminoguanidine bicarbonate was separated as fine white precipitate. Precipitate is filtered, washed with small amount of cold
water and dried. Product is white microcrystalline powder, yield is 49.1g (85% based on hydrazine, Note №4).
Notes:
1. This pH range can be achieved without adding sulfuric acid – by adding components in
corresponding rate, however it can also be justified by addition of acid. Addition can be carried out in portions, adding new portion, stirring for
1-2 minutes then adding new one e.t.c. Addition should not produce any gas or significant odor, if such are present – your cyanamide is impure and
yield be lowered. Use of cyanamide contaminated with alkali metals on this step will lead to formation of hydrogen cyanide witch will be noticeable by
odor, while cyanamide significantly oxidized by air during production (e.g containing carbonate) will form a lot of bubbles of carbon dioxide gas.
2. Total amount of sulfuric acid, consumed during whole reaction process is equivalent to 12-15 ml
of concentrated 96% H2SO4. It should be noted that not uniform addition of sulfuric acid will cause high local concentrations of H2SO4 and formation
of solid/gases, so if H2SO4 is added by portions for example from syringe, addition must be accompanied by vigorous stirring to prevent loss of
reactants.
3. Precipitated calcium sulfate consist from particles of relatively large size and filtration
proceeds with ease. Total amount of water used for washing is not critical however 200 ml is reasonable amount. One can notice that filtrate obtained
has distinctive yellow coloration common for solutions of aminoguanidine salts.
4. Product yield can vary depending on quality of starting cyanamide and from care with witch
correct pH ranges are controlled.
Photos:
1. Left photo shows reagents – calcium cyanamide and hydrazine sulfate used for reaction, second
photo is reaction mixture in the middle of reagent addition, third one is heating stage, last photo shows reaction mixture with calcium sulfate
precipitate, obtained after heating period.
2. Left photo shows filtration from calcium sulfate, second is filtrate produced –note that this
yellow color is common for solutions of aminoguanidine salts, third photo is precipitate of aminoguanidine bicarbonate formed after addition of sodium
bicarbonate, last photo is white pure aminoguanidine bicarbonate produced by the process.
Chapter 2 – Aminoguanidine, nitroguanidine route
Step №1. Synthesis of guanidine nitrate from urea and ammonium nitrate.
Properties. Guanidine nitrate [C(NH2)3]NO3 is colorless plates (in presence of impurities
yellowish) with density 1.44 g/cm3 and melting point 214.2C. Compound is stable to boiling in water and on melting, decomposition starts only at 270C.
Soluble in water (13% at 20C, 32% at 55C and 128% at 90C) and ethanol (11.5% at 78C).
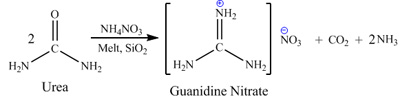
Procedure. Guanidine nitrate can be prepared by reaction of urea with ammonium nitrate in molten
state in presence of powdered silica gel catalyst at temperature about 160C. Reaction equation:
Reactor is open 2 liter stainless steel container placed on oil bath heated by hotplate and equipped by mechanical
stirrer. Stirrer was made from small 9V motor (from old tape recorder) with shaft made from radio antenna section and stirring arms made from
stainless steel, all open surfaces were covered by P.T.F.E lacquer (Note #1). Reactor is filled with 340g of ammonium nitrate (Note #2), 200g urea and
80g of tempered dry silica gel (Note #3) and heated up on oil bath to 195C. Mixture melts and then stirring is applied for total 2 hour sitting
period, then another 120g of urea is added and mixture is allowed to sit for 2.5-3 more hours at this temperature (urea can not be added at start in
single portion because it will lead to formation of triazine compounds). After and of this period mixture is cooled, mixed with 250 ml of water and
boiled, filtered hot to remove silica gel, then material on filter washed with 150 ml of boiling water. Filtrates are combined and cooled to 0C,
precipitated guanidine nitrate is filtered and recrystallized from minimal amount of water. Yield is 190-210g of pure recrystallized product (65-70%
theoretical).
Hazard note:
Process of melting urea with ammonium nitrate is related with explosion hazard, explosion much likely to occur then
mixture contain both reagents in zero oxygen balance proportion or if temperature is extended over proposed limit. Described process have been carried
out by people from local forums many times without any explosions, however one should consider potential possibility of such then performing this
process and take to account all necessary safety precautions.
Notes:
1. Experimentation in performing the process without stirring (mixing was applied only once –
then adding second crop of urea), shown that yield is lowered by 2-3%. However stirring is efficient in prevention of local overheat, lowering
explosion hazard.
2. Using dry reagents is essential to reduce foaming, especially on first stage.
3. Used silica gel was prepared from liquid glass (sodium metasilicate) using the following
procedure. Commercially available liquid glass is diluted 2-3 times, then 10-15% sulphuric acid is added. Solution forms non fluid galantine (it is
essential that acid should be added in one portion, and solution should thicken, if H2SiO3 precipitate is formed, solutions must be more diluted and
mixed more efficiently) witch is placed to the cold freezer for 2 days. On defrost galantine disperses to fine powder, witch is filtered, carefully
washed and tempered to dryness. This silica gel was reused in guanidine nitrate process for 7 times, without loosing activity. Regeneration of silica
gel was preformed by washing thoroughly with 1 liter of hot water and tempering.
Photos:
1. Reaction vessel and pure guanidine nitrate, made by method described above.
Step №2. Preparation of nitroguanidine by dehydration of guanidine nitrate.
Properties. Nitroguanidine C(NH)(NH2)NHNO2 is colorless or slightly yellowish fiber like crystals
with monocrystalline density 1.71 g/cm3 and melting point 232C (with decomposition). Nitroguanidine is soluble in common organic solvents, good
soluble in water (0.44% at 30C, 7.5% at 100C) and perfectly soluble in alkaline solutions. Heat of formation is -893 kJ/kg, heat of explosion 3220
kJ/kg (liquid water) and 2876 kJ/kg (water – gas), trauzl bomb test gives 305 cm3 expansion, volume of detonation products – 1075 l/kg, oxygen
balance -30.7%, 54% mass nitrogen. Nitroguanidine is typical “cold” explosive with low explosion temperature, while explosion workability is
slightly higher then TNT. High crystal density and low molecular mass of explosion products results in relatively high detonation velocity (7650 m/s
at 1.55 g/cm3, 8200 m/s TMD), so nitroguanidine can be referred as high explosive. Sensitivity to external stimulus is quite low (no detonation on
usual impact and friction tests).
Derivatives.The silver derivative, CH3N4O2Ag, is precipitated when barium hydroxide is gradually
added to a warm aqueous solution containing nitroguanidine and silver nitrate in molecular proportion; it is a colorless compound, almost insoluble in
water, but readily soluble in acids, ammonia, and ammonium salts ; it separates from a hot solution of ammonium nitrate in microscopic needles. It
turns yellow when treated with alkalis, and darkens on exposure to the air or on prolonged washing with water; it has an alkaline reaction, and
explodes when heated. A yellow precipitate, which seems to have the composition CH2N4O2Ag2, is formed when soda or barium hydroxide is added to a
solution of nitroguanidine in ammoniacal silver nitrate; it is very hygroscopic, and decomposes very readily when dried at a moderate temperature. The
nitrate, CH4N4O2*HNO3, crystallizes from hot concentrated nitric acid in nacreous plates, and melts at 147°C. The hydrochloride, CH4N4O2*HCI,
crystallizes in plates or prisms. When nitroguanidine is treated with soda and zinc-dust, and then with a solution of a ferrous salt, a beautiful red
coloration is produced; attempts to prepare alkyl and acidyl derivatives of nitroguanidine were unsuccessful.
Procedure.Nitroguanidine can be prepared by dehydration of guanidine nitrate with concentrated
sulphuric acid, followed by dilution by water and filtering:
Aging period depends from temperature, at 40C 5min period is suitable, 30C requires 10 min and 20C require 30 min of
aging. Dehydration uses 2.5-3 times mass quantity of concentrated H2SO4. 500g of 93-95 sulphuric acid is placed on water/ice bath and cooled to 10C,
then guanidine nitrate (~200g) is added by portions with stirring (~5-10g at a time) in such rate that at the end of addition temperature should reach
30C (temperature rises relatively slowly, but cooling is still required), then mixture is aged for 10 minutes and poured into 300g of water/ice
mixture. Fine precipitate of nitroguanidine is filtered, washed with strongly diluted water solution of ammonia, with cold water and finally with
alcohol. Yield is about 142g (85% theory).
Photos:
Crude nitroguanidine made by method above and product recrystallized from water.
Step №3. Synthesis of aminoguanidine by reduction of nitroguanidine with zinc in acetic acid.
Properties. Properties of aminoguanidine and some simple derivatives vere described in
corresponding part of previous chapter and here are omitted.
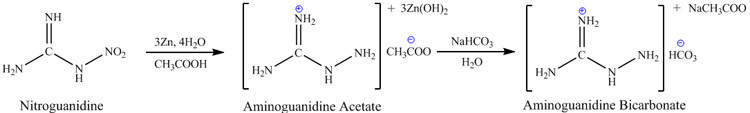
Procedure. Aminoguanidine can be prepared by many routes, for example by action of hydrazine on
cyanamide or methylisothiourea and by reduction of nitroguanidine by hydrogen. Probably the best practical method of reduction is reduction of
nitroguanidine by zinc dust in acetic acid, since it does not require additional catalysts and is very simple. This method offers 15.6g of
aminoguanidine-bicarbonate from per 20g of nitroguanidine (60% from theory):
20g (0.19 mol) of nitroguanidine, 68g (1.04 mol) of zinc dust and 6 ml water are mixed in mortar and rubbed to thick
paste. Reaction weasel (thin-wall glass) is filled with 12 ml of glacial acetic acid + 12 ml water and placed to ice/water bath, then cold
nitroguanidine/zinc paste is added in small portions with gentle stirring (Note №1). Addition rate is adjusted to hold reaction temperature
within 5-15C, care should be taken not to allow temperature to rise to 35C even for a single moment. If mixture becomes too thick or temperature rise
to rapidly pieces of ice are added (total 90g). Paste addition takes about 3-4 hours, after this reaction mixture is allowed to sit in ice/water bath
for 1 more hour, then mixture is removed from bath and is allowed to heat slowly to room temperature, final mixture volume is about 130-140 ml (Note
№2). After 1 hour at room temperature mixture is placed on water bath, heated to 25 and stirred for 30 minutes, then to 32-35C and stirred for
30 more minutes and finally heated to 40C and stirred at this temperature for 15 minutes (Note №3). Then reduction is complete solution is
immediately filtered on vacuum funnel, taking effort to filter solid product to dryness (note #4). Solid is washed with 90 ml of water, filtered and
discarded, filtrates are combined, 18.5g of ammonium chloride is added, and mixture is stirred until all ammonium chloride is dissolved (note #5), and
on continuous stirring 20.2g of soda is dissolved in reaction mixture. Several minutes from addition aminoguanidine-bicarbonate starts to precipitate.
Mixture is placed to cold place and allowed to stand for one night, and precipitated aminoguanidine bicarbonate is filtered, washed with ethanol and
dried. Yield is 15.6g (60% theory) of aminoguanidine bicarbonate in form of dense white powder, melting with decomposition at 172C.
Notes:
1. Addition of several first crops of paste is accompanied by virgeous reaction and strong heating.
On addition of later portions of paste reaction begins to cool down, and strong heating is slowly ceased. Then reaction becomes calmer larger portions
of paste could be added, while taking care to not allow to fast temperature elevation. After addition is near completion reaction slows down and
exotherm occurs not permanently, but some time after the addition. If temperature is rising too fast due to inefficient stirring of too thick mixture
or if added paste portion was too large it is convenient to add some crushed ice to lower mixture temperature and make stirring easier.
2. To rapid heating of reaction mixture should be avoided, because reaction proceeds faster and
becomes exothermic. It should be remembered that aminoguanidine is unstable to heat and to much heat will cause decreased yield of product.
3. Precise timings may be varied from run to run. To determine completion of reduction it is
convenient to use qualitive test for nitroguanidine witch is preformed as follows. 3 drops sample of reaction mixture is added to test tube with 5 ml
of 10% sodium hydroxide, and 5 ml of fresh saturated solution of Mohr's Salt (Ammonium iron(II) sulfate, or just mixture of NH4SO4 + FeSO4 solutions)
is added. Red coloration shows presence of unreduced nitroguanidine. If reduction is complete same test results in formation of greenish precipitate
without coloration of solution. As soon as test shows complete reduction, reaction mixture should no longer be heated and must be filtered as soon as
possible.
4. Reaction mixture is quite thick and filters not readily. To force filtering to dryness, solid on
filter should be trampled down with spoon in effort to fill all cracks forming while solid changes volume to prevent easy entry of air through the
filter funnel. Good filtered solid should be almost dry. Filtering after later washing of precipitate goes without any difficulties.
5. Presence of ammonium chloride prevents joint precipitation of zinc salts then sodium
hydrocarbonate is added to precipitate aminoguanidine as weak soluble bicarbonate. If on this stage solution is not transparent it should be filtered.
Ammonium compounds transform zinc salts to soluble ammoniacal complexes, allowing to precipitate pure aminoguanidine bicarbonate.
Photos:
1. Source products – zinc and nitroguanidine and thick paste used in reaction.
2. At beginning of addition reaction mixture tend to heat vigorously, so temperature should be
controlled carefully. During this period paste should be added in small portions with efficient stirring, fortunately thickness of mixture is still
low so this could be done with relative ease.
3. While new portions of paste are added, mixture becomes thicker and heat exchange becomes more
and more difficult, local overheating is possible. If stirring is not efficient enough temperature difference in some places of mixture can be as high
as 10-15C. Stirring and heat exchange can be cured by adding portions of crushed ice.
4. Then paste addition is complete, reaction mixture is allowed to stand at room temperature,
giving time for exothermic reaction to finish and heated in water bath as described above. To keep temperature in required interval it is convenient
to use hot water stream with temperature 2-3C higher then required.
5. Left photo shows qualitive reaction for nitroguanidine. Then few drop sample of reaction mixture
is added to alkaline solution of iron (II) salt or iron-ammonium complexes nitroguanidine forms beautiful red coloration, while aminoguanidine in
absence of nitroguanidine forms greenish precipitate shown on right photo.
6. Reaction mixture is quite thick and can not be filtered easily, it is highly recommended to use
wide vacuum funnel to ease filtering. Filtrate has characteristic yellow color showing presence of aminoguanidine salt and also contains impurities -
guanidine and zinc salts.
7. To exclude cooperative precipitation of zinc salts on addition of soda, required amount of
ammonium chloride is added in order to bind zinc in soluble zinc-ammonium complexes. After addition of ammonium chloride and soda aminoguanidine
bicarbonate begins to precipitate in form of dense white powder.
Chapter 3 – Transformation of aminoguanidine to 5-aminotetrazole.
Properties. 5-Aminotetrazole was first prepared by Thiele, by diazotation of aminoguanidine with
sodium nitrite in hydrochloric acid environment. 5-Aminotetrazole crystallizes from water solution in the form of the monohydrate, which are colorless
prisms or leaflets, losing water above 100°С and melting with decomposition at 200-203°С. 5-Aminotetrazole is badly soluble in alcohol
and more readily in ether. It is also soluble in water solutions of bases and strong acids, and has good solubility in hot and poor solubility in cold
water. 5-aminotetrazole's heat of combustion is 246.2 kcal/mol, standard enthalpy of formation is -49.7 kcal/mol. 5-Aminotetrazole shows weak acid
properties, and in the anhydrous state is extremely hygroscopic. It is stable to heat, and its dissociation constant is about 1*10-4. Besides its acid
properties, upon reaction with strong mineral acids 5-aminotetrazole can act as base, as can many organic amines. In general 5-aminotetrazole acts as
an amphoteric substance, with behavior similar to that of amino acids. The high chemical stability of the tetrazole ring in addition to fact that
substituents on the tetrazole ring are usually entered during ring formation, leaves the amino group as the only reasonable target for chemical
manipulations. The amino group of 5-aminotetrazole has all the common properties of that functional group, and can be related in chemical behavior to
the amino group of aniline.
5-Aminotetrazole forms salts with metallic cations, some of them are explosive. The cobalt salt Co(CN5H2)2*H2O,
exists as pink water soluble crystals which explode upon heating to 228°С. The nickel salt Ni(CN5H2)2*H2O exists as blue water soluble crystals
which explode upon heating at 290°С. The lead salt Pb(CN5H2)2 , exists as colorless crystals which deflagrate upon heating to 303°С. The
mercury salt Hg(CN5H2)2 exists as white water insoluble crystals, they explode when dropped on a hot plate heated to 256°С. The Hg salt's shock
sensitivity in the lead weight test is 50% explosions using a 2.5 kg weight and 38 cm drop height. The copper salt Сu(CN5H2)2*H2O exists as
green very slightly water soluble crystals. A 2.5 kg weight with an drop height of 68 cm gives 50% of explosions when the copper salt is tested and it
deflagrates when heated to 164°С. Aminotetrazole in the presence of copper sulfate and sodium acetate produces a green amorphous precipitate,
which can be used as a diagnostic test for aminotetrazole. This precipitate is insoluble in acetic acid, and is soluble in hydrochloric acid.
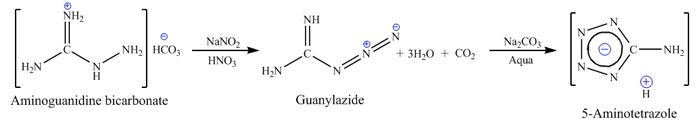
34g (0.25 mol) of aminoguanidine bicarbonate is added to 217 ml of 15% nitric acid (0.561 mol), and mixed until
evolution of carbon dioxide is stopped and resulted aminoguanidine nitrate is fully dissolved in solution. Yellow transparent solution is diazotized
by slow addition of 17.2g sodium nitrite (0.25 mol) in 35 ml of water. Addition is accompanied by stirring, and temperature during all addition period
is kept between 20-25°С by using water bath if needed (note #1). After completion of reaction the diazotation mixture is allowed to sit for 20
minutes at room temperature, and 29g of sodium carbonate is added (or 46g of sodium bicarbonate). Mixture is then heated on a water bath and refluxed
for 4 hours. The solution is then neutralized by 30% sulphuric acid to pH=4, cooled to room temperature and allowed to sit over night (note#2). The
precipitated crystals of 5-aminotetrazole monohydrate are filtered, washed with cold water and dried. Yield is about 70-74% based on aminoguanidine.
Notes:
1. Diazotation proceeds smoothly with little exotherm. If reaction mixtures begins to foam (this is
the result of decomposition of nitrous acid), mixture must be stirred until form is settled before adding new a portion of nitrite solution. If
reaction is carried put in the right way, it takes time about 10-15 minutes and proceeds with negligible evolution of nitrogen oxides.
2. 5-Aminotetrazole has an affinity to form supersaturated solutions. Often it does not begin
crystallization even after full cooling, in that case crystallization should be assisted by introduction of a seed crystal, or by rigorous friction of
a glass rod on side wall of reaction vessel (below liquid). Crystallization is generally fully complete after 12 hours from start.
Photos:
[Edited on 21-8-2010 by Engager]