| Pages:
1
2 |
mario840
Hazard to Others

Posts: 229
Registered: 20-1-2010
Member Is Offline
Mood: No Mood
|
|
N-Butylamine
Hello
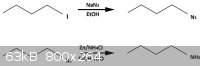
I have successfully prepared n-butylamine. At first i made n-butyl azide from n-butyl iodide (buy) and sodium azide in methanol (procedure from
Organics Functional Group Preparations by Stanley r. Sandler instead bromide i used iodide also it's better leaving molecule). Second step is
reduce azide to primary amine , i started search for easy and convinient method and found reduction with Zn/NH4Cl (Reduction of Azides to Amines
or Amides With Zinc and Ammonium Chloride as Reducing Agent). So i started my last step according to this method i should get about 90% yield but
i got +- 80% , this is fantastic because some product lost while i destill it to purify and this was my first attempt. This method is simple, very
cheap (some method need Pt  ) and quick , high yielding ( i reflux for about 30
- 45 minutes to sure that reaction is complete). This reaction is basic for preparation of primary amines from azides but the reduce method is very
good and not usual. ) and quick , high yielding ( i reflux for about 30
- 45 minutes to sure that reaction is complete). This reaction is basic for preparation of primary amines from azides but the reduce method is very
good and not usual.
References :
"Organic Functional Group Preparations" Stanley R. Sandler
"Reduction of Azides to Amines or Amides With Zinc and Ammonium Chloride as Reducing Agent" W. Lin, X. Zhang, Z. He, Y. Jin, L. Gong, A. Mi Synth.
Commun. 32(21), 3279-3284 (2002)
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
It is nice that it worked well for you, but you might consider sharing the experimental as well, so that other members interested can learn something
from your experience. In it you should address the key issues of this preparation, namely the issues of product isolation and characterization. How
did you separate n-butylamine from ethyl acetate? You give the yield, which leads us to believe that you already characterized the product. So how did
you confirmed the identity and purity of the product? n-Butylamine is a liquid at standard conditions, so to characterize it in an amateur setting you
would have to derivatize a sample and measure its m.p. (have you checked the m.p. of the hydrochloride?).
For amateur chemists, it is not so much the reaction itself that is an issue, but the isolation and characterization, so you would be most helpful if
others could learn from your problem solving approach.
|
|
|
mario840
Hazard to Others
Posts: 229
Registered: 20-1-2010
Member Is Offline
Mood: No Mood
|
|
1. Adding all together in the same ratio of moles like in procedure reduction only multiply everything by x10
2. Heating under reflux at first 30 minutes but i heat just for 15 minutes to sure that reaction is done, so 45 minutes is just fine
3. I do NOT add ethyl acetate !!!! only dietyl ether , n-butylamine and EA have almost the same boiling point so i need something "lower" boiling ---
i choose Et2O
4. I filter solution and wash with brine and dry over anh. MgSO4 (couple hours)
5. Evaporate solvent (steam bath) i don't need reduce pressure beacuse i use ether (earlier i smeel stinky dead fishy odor but now it's stronger plus
irritating but not like ammonia)
6. Residue is destilling and collecting everything 75C - 79C
7. For sure me that is n-butylamine i made Hinsberg test , so i don't have gas chromatograph but i'm sure that is n-butylamine
|
|
|
mario840
Hazard to Others
Posts: 229
Registered: 20-1-2010
Member Is Offline
Mood: No Mood
|
|
Also i want to add i thought about Gabriel synthesis wich also give me n-butylamine but yields will for sure lower and price in my side phthalimide
are crazy
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
Thanks for sharing.
If you are still interested in n-butylamine synthesis, you can try the Delepine reaction and compare the results (n-butyl iodide is a primary alkyl
halide, thus amenable for this type of amine synthesis and hexamine is OTC in most parts of the world).
|
|
|
mario840
Hazard to Others
Posts: 229
Registered: 20-1-2010
Member Is Offline
Mood: No Mood
|
|
Thanks men for that reaction. I read about this Delépine reaction and seems to be cheaper , only need n-butyl iodide and urotropine , chloroform and
ethanol with conc. HCl (these all is still cheaper of NaN3, Zn and NH4Cl etc.). Also i like in these reaction that forming quaternary ammonium salt is
easily to filtrate ("...moderate colubility in water (5%), less in alcohol (0.5%) and less still in chloroform (0.08%) and ether...), so we
don't need playing with destillation , great plus is that amine (n-butylamine) will be end in HCL form, minus is that this synthesis took longer but i
can live with that. I definitely try this procedure. Also they write alkyl iodide react with reasonable speed while bromide and chloride much longer
so it's good for me.
|
|
|
g3ovn
Harmless
Posts: 12
Registered: 8-11-2010
Member Is Offline
Mood: No Mood
|
|
Sorry my ignorance, but n-butylamine hydrochloride can't be made from n-butyl nitrite reduction with Fe/HCl?
|
|
|
smuv
National Hazard
Posts: 842
Registered: 2-5-2007
Member Is Offline
Mood: Jingoistic
|
|
No. With nitrobutane you may have something, but with butyl nitrite, you would probably only get butyl chloride, butanol and maybe some butane, under
the conditions you specify.
"Titanium tetrachloride…You sly temptress." --Walter Bishop
|
|
|
mario840
Hazard to Others
Posts: 229
Registered: 20-1-2010
Member Is Offline
Mood: No Mood
|
|
n-butyl NITRATE and reduction with zinc/ammonium formate with about 50 - 60% yield, but making n-butyl NITRATE is hard and expensive there are cheaper
methods
|
|
|
mario840
Hazard to Others
Posts: 229
Registered: 20-1-2010
Member Is Offline
Mood: No Mood
|
|
Also i made delepine reaction :
-with alcohol as solvent it doesn't work (or have to stay for 42 days to get salt of hexamine TOOOO LONG !)
-it only work in chloroform (12 HOURS boiling - TOO LONG ALSO !)
-filtrate salt and make slurry in alcohol add conc. HCl to disolve salt and boil 30 minutes , chill in ice and NH4Cl get out from solution
-filter , evaporate to dryness and get n-butylamine*HCl, but massive impure
-disolve in water add 45% NaOH and extract with ether
-evaporate ether and get amine
-for puryfy destill at 77-78C
THIS method is usless , only it work at good speed to 3 atoms chain , so n-propylamine we will get in 5 hours boiling
|
|
|
g3ovn
Harmless
Posts: 12
Registered: 8-11-2010
Member Is Offline
Mood: No Mood
|
|
Oh, thanks for sharing it!
I think reacting butyraldehyde with ammonia, and then reducing the imine will work also...
|
|
|
mario840
Hazard to Others
Posts: 229
Registered: 20-1-2010
Member Is Offline
Mood: No Mood
|
|
Maybe Gabriel synthesis work faster then Delepine with 4 carbon chains, phtalimide is expensive but make easily from anhydride and ammonia
|
|
|
smuv
National Hazard
Posts: 842
Registered: 2-5-2007
Member Is Offline
Mood: Jingoistic
|
|
| Quote: | | n-butyl NITRATE and reduction with zinc/ammonium formate with about 50 - 60% yield |
Really? Do you have a ref, I would not expect this to work. Again with nitrobutane, yes but not with butyl nitrate.
"Titanium tetrachloride…You sly temptress." --Walter Bishop
|
|
|
mario840
Hazard to Others
Posts: 229
Registered: 20-1-2010
Member Is Offline
Mood: No Mood
|
|
This is simple procedure , it work with many nitroalkanes :
Zinc-Catalyzed Ammonium Formate Reductions: Reduction of Nitro Compounds
By D. Gowda, B. Mahesh, & G. Shankare, Ind. J. Chem. Sect. B, 40, 75-77 (2001)
there are many others method like : zinc/hydrazinum formate and magnesium/hydrazine formate but these require inert atmosfere like nitrogen , the
zinc/NH4COOH or if you read that also 90% formic acid work ! don't need nitrogen
|
|
|
mario840
Hazard to Others
Posts: 229
Registered: 20-1-2010
Member Is Offline
Mood: No Mood
|
|
you have right only with nitrobutane
|
|
|
g3ovn
Harmless
Posts: 12
Registered: 8-11-2010
Member Is Offline
Mood: No Mood
|
|
I made from phtalic anhydride and urea.
50g phtalic anhydride and 10g urea. Place it in an oil bath at 130-145°C it first starts to
melt, then it "boils" and condenses the phtalic anhydride in the flask.
Suddenly it froths up like 3 times (like in Vogel). Then i take it out the heat, waited a little and put 80mL of dH2O, filtered, and washed with cold
water (solubility 1g/1L) until i coldn't smell any more ammonia from the water. Place it in water bath for 2h. The yield was 45g. It can be
recristallized from lots of methanol, Vogel says 1200mL for 86g.
Store it in the dark.



[Edited on 26-6-2011 by g3ovn]
[Edited on 26-6-2011 by g3ovn]
|
|
|
jon
Hazard to Others
Posts: 459
Registered: 11-1-2006
Member Is Offline
Mood: paranoid distrustful apprehensive
|
|
sir, i am very interested in your work with azides.
on paper they look dodgy as hell.
can you offer any real world advice on the handling of organic azides?
i understand they are shock/friction sensitive compounds very labile and reactive.
|
|
|
mario840
Hazard to Others
Posts: 229
Registered: 20-1-2010
Member Is Offline
Mood: No Mood
|
|
Theory doesn't sometimes work in practise, n-butyl azide have relativity high boiling point (about 107 C) so basic safety "gear" like gloves, goggles,
clothes to change, work in fumehood or outside works just fine if you have gas mask it's good for you. When you synthsise compound (azide) do not
"overheat"; boiling temperature of solvent which you use is just fine (best methanol, ethanol is expensive of course anhydrous like methanol), the
next advice is when you destill alkyl azide try to get as close as temperature of boiling compound (not higher) beacuse better is steady and slow
destillation then explosion, the best option is destill behind a safety barricade like they write in book, but i doesn't use that. I destill many
times much more toxic compund than that so try to not demonize too much some chemical. In file i give you method of preparation of alkyl azides in
microwave in short time, they use only temp. 100 C !! so try also do not "overheat", like i said in traditional way of synthesise it boiling point of
solvent is just fine. Butyl azide and methanol form a azeotrope (b.p 60 C) so you must have very good reflux condenser, but how to separate them they
write in book.
Attachment: microwave azides.pdf (160kB)
This file has been downloaded 2177 times
|
|
|
jon
Hazard to Others
Posts: 459
Registered: 11-1-2006
Member Is Offline
Mood: paranoid distrustful apprehensive
|
|
i guess for small scale working without a blast sheild is ok but preparative scale i think it would be a must.
personally i have been cut to shreads with exploding flasks, it was'nt very fun.
|
|
|
g3ovn
Harmless
Posts: 12
Registered: 8-11-2010
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by mario840  | 1. Adding all together in the same ratio of moles like in procedure reduction only multiply everything by x10
2. Heating under reflux at first 30 minutes but i heat just for 15 minutes to sure that reaction is done, so 45 minutes is just fine
3. I do NOT add ethyl acetate !!!! only dietyl ether , n-butylamine and EA have almost the same boiling point so i need something "lower" boiling ---
i choose Et2O
4. I filter solution and wash with brine and dry over anh. MgSO4 (couple hours)
5. Evaporate solvent (steam bath) i don't need reduce pressure beacuse i use ether (earlier i smeel stinky dead fishy odor but now it's stronger plus
irritating but not like ammonia)
6. Residue is destilling and collecting everything 75C - 79C
7. For sure me that is n-butylamine i made Hinsberg test , so i don't have gas chromatograph but i'm sure that is n-butylamine |
I didnt got it, in the book you are refering to says 24h in a steam bath for n-butyl bromide!
I think at least 6h for the all the iodide the react. Where do you saw 30 min??
| Quote: | | distilled behind a safety barricade to yield 40.0 gm (90%) of n-Butyl azide, b.p. 106.5°C |
Just for the warnings i think that's too much a risk, even more for 24h of heating (or 6h)! I think there are more safer, quicker and less expensive reactions... i mean i saw 1L of AR n-butylamine for $85, doing like 200mL of it
by that reaction will cost me about that.
But thank you for sharing it! I think i will be trying Gabriel synthesis with n-butyl bromide, potassium phtalimide and sodium hydroxide.
Attachment: 35932599-Organic-Functional-Group-Preparations-Volume-2-Stanley-R-Sandler-and-Wolf-Karo-1971.pdf (1.6MB)
This file has been downloaded 1924 times
[Edited on 27-6-2011 by g3ovn]
|
|
|
smuv
National Hazard
Posts: 842
Registered: 2-5-2007
Member Is Offline
Mood: Jingoistic
|
|
Not a problem. A buddy in my lab used to work with azides on a large scale all the time. No great precautions were taken. Never a mishap. I have
only prepared a couple of azides (both ~10g); no great precautions, no mishaps.
Don't sweat it.
"Titanium tetrachloride…You sly temptress." --Walter Bishop
|
|
|
mario840
Hazard to Others
Posts: 229
Registered: 20-1-2010
Member Is Offline
Mood: No Mood
|
|
g3ovn read more carefully , this is procedure for REDUCING alkyl azide to primary amine ! how to make azide is write in book, Gabriel synthesis is
this what i'm thinking about too, delepine is only good for allyl, aryl short carbon chain aliphatic IODIDE so making allylamine is good choice (when
i will buy allyl alcohol and HBr acid i will definitly try this one), remeber when you will making Gabriel the final step is better add hydrazine in
ethanol reflux that for 1 hour, cool and then add HCl because insoluble phthalhydrazide separetes out, while amine*HCl is in solution that make easily
to purify amine of course only NaOH will also work ; and don't forget post your results
|
|
|
jon
Hazard to Others
Posts: 459
Registered: 11-1-2006
Member Is Offline
Mood: paranoid distrustful apprehensive
|
|
thanks for the real world advice people.
if your looking for advice on hydrobrominations i've done it hundreds of time on a certain essential oil with 92% yield everytime.
the idea is to make the hbr insitu in acetic acid from sodium bromide and h2so4.
it's easy the less you mess with it the better the result.
i suspect some of my synapses are'nt there anymore as a result.
p.s.
i noticed a burette clamp this is bad lab technique.
use a 3 prong next time, you get better extention and grip that way.
[Edited on 27-6-2011 by jon]
[Edited on 27-6-2011 by jon]
|
|
|
smuv
National Hazard
Posts: 842
Registered: 2-5-2007
Member Is Offline
Mood: Jingoistic
|
|
A nice thing about the gabriel is the intermediate substituted pthalimides are easy to purify and generally recrystallize nicely from lots of solvent
systems. You can put a crude halide into the gabriel and get out a pure amine. Also, hydrazine isn't necessary for simple amines, extended
saponifaction gets the job done (and is how the reaction was originally preformed).
@jon doesn't seem like they were all needed (synapses).
[Edited on 6-27-2011 by smuv]
"Titanium tetrachloride…You sly temptress." --Walter Bishop
|
|
|
mario840
Hazard to Others
Posts: 229
Registered: 20-1-2010
Member Is Offline
Mood: No Mood
|
|
Now i regred that i tried delepine reaction ,i should try gabriel, much shorter reaction time for longer chain carbons , it's done even with chloride
(delephine reacts for months in room temperature with chlorides) iodide react of course much faster , reaction can be done in dimethylformamide or
ethanol (delepine only in chloroform - the first step), there are couple substitutions for phtalimide in this reaction for example : sodium
diformylamide is easily prepared from sodium, methanol, formamide (i got everything) but still phtalimide is better option (cheaper)

|
|
|
| Pages:
1
2 |








