| Pages:
1
..
10
11
12
13 |
OneEyedPyro
Hazard to Others

Posts: 280
Registered: 7-10-2015
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by ManyInterests  | | Quote: Originally posted by OneEyedPyro | Brining up the temperature too much toward the end of nitration isn't necessary with PETN, it's rather easily brought to full nitration by bringing it
up to 25-30C in warm water for the last half an hour.
In my experience PETN is fairly tame and not prone to runaway IF constantly/thoroughly stirred in a cold water bath and the PE additions are done in
a few steps with a couple minutes in between to make sure the temp isn't climbing which for me it never has.
I would guess the NO2 evolving when you heated it was likely decomposition of your PETN, that's probably where your yields went.
|
Duly noted. Next time I will simply allow the PETN to stay at room temperature for a while before simply crashing it in a large amount of ice cold
water.
I will also use an excess of around 75% nitric acid and go as strong as 90%. I am hoping to have the yield that others have had (2.2g PETN per 1g PE)
than the poor yield I got.
I will also time it and try to not to worry too much about the yellowing. It is probably not as bad as I made it out to be. |
A lot of procedures involve heating to around 40-45C for 20 minutes or so before crashing it out, I've tried it and found that 30C works about the
same with less fumes to deal with. The yellowing may be indicative of impurities due to dehydration/ooxidation.
If you run a small batch at just above room temp you'll find decent yields, maybe not mid 90% but still acceptable.
|
|
|
DennyDevHE77
Hazard to Others
Posts: 162
Registered: 15-9-2014
Member Is Offline
Mood: No Mood
|
|
So, as I understand it you used the sulfur-nitrogen method of nitration in one stage, but you used the proportions for 70% nitric acid. You should
realize that too strong sulfur-nitrogen nitro mixtures oxidize PETN much easier than nitrates.
Correct nitration should be carried out as follows: Pentaerythritol is added to the prepared nitro mixture cooled to 10°C, the temperature is not
allowed to rise above 15°C. If this limit is exceeded, thermally unstable compounds are formed, which will show themselves on further heating. After
the addition of all pentaerythritol, give a rest of 5 min, continuing stirring.
Then the beaker with PETN is placed in a water bath with warm water. It is necessary to gradually raise its temperature to 25-30°C. The water in the
water bath is then replaced by very hot water and the main heating to 50°C is started. When the temperature of PETN reaches 50-52°C feel free to
take the beaker out of the water bath, and return it when the temperature drops below 50°C again. At this stage, the sulfo groups in PETN are
replaced by nitro groups, this is called the transesterification reaction. It starts at +35°C but reaches an acceptable rate at +50-60°C. So no 25-30°C, it is
not enough and therefore useless, or heat normally, at least to 50°C or pour into a bucket of water without heating at all, after a 5-minute soaking
time. For PETN by this method, 20 min at 50°C is sufficient. From personal experience I can say that this is the most calm temperature, during all 20
min light yellow vapors are emitted, and no more. I not infrequently increased the temperature to 55-60°C, then more vapors are released and the
reaction mass begins to bubble in 15 min. OneEyedPyro is right above, saying that PETN is not prone to escape. If you are concerned about nitrous
oxide vapors and yellowing of the mixture, you can just blow into the beaker and blow out the nitrous oxide and the mixture will be white)))
The real sign of escape is a strongly bubbling, almost boiling mixture, that's when you can safely pour it into a glass of water. And I'll tell you
right away that I had a few runaways a long time ago, when I chickened out and just did not dare to take a glass and pour it into the water. It began
with a bright red fire in the center of the boiling mixture, after which the whole mass caught fire, with the release of large quantities of NO2, and
since I did not use in my school days heat-resistant utensils, then when burning nitro-mixture glass burst in the water bath, the mixture was diluted
with water, and the burning stopped. it remained only to ventilate the apartment from nitrogen dioxide. There was never an explosion in 3 runs.
However, with your mixture with 90% nitric acid I am almost sure that oxidation could start abruptly at 50°C.
And you really can not heat your PETN (but this will require longer cleaning), the yield will be on average 70-80%, or it is possible to heat for a
short time, as at 50°C for 7-10 min of heating the yield already becomes about 90%, at full transesterification the yield is 92%. I often produce
PETN, with 100g of pentaerythritol I consistently get ~220g.
In general, if you have 90%, you would be better off immediately nitrating with pure nitric acid, getting the same yield of 90%, but saving yourself
from lengthy cleaning. Just in case, I will point out that nitration is carried out with 90-99% nitric acid at the rate of 100 g of acid per 20 g of
pentaerythritol at a temperature of 20°C. This method is convenient for cleaning.
You see, PETN obtained by a two-stage or one-stage method through a nitro mixture is contaminated with mixed sulfoesters, which reduce its power and
stability, since previously PETN was produced in two stages industrially, this issue is generally well studied. So, especially if you did not heat the
mixture, you need to pour 9-10 parts of a 1% aqueous solution of sodium bicarbonate, and boil your PETN until it stops foaming. Instead of baking
soda, you can use, for example, magnesium oxide or sodium carbonate, but here it is better to keep the temperature at 90°C, which is less convenient;
at 100°C there will be slight decomposition.
Then recrystallization from boiling acetone with soda. And yes, the neutral solution of PETN in acetone, in principle, has a yellowish color, and even
more so with traces of sulfoesters, at this stage they are completely destroyed. In the photo below I dropped what my PETN looks like in acetone,
usually by the way it is even more yellow, here there is simply an excess of acetone.
I’ll also post a photo of what PETN looks like at 51°C after 10 minutes of heating; in this photo, the glass was removed from the water bath to
prevent further heating. As you can see there are only small yellow vapors present here.
And the same advice) do not try to immediately filter PETN after pouring the nitro mixture into water, PETN obtained using the sulfur-nitrogen method
is very fine and without vacuum filtration, its filtration takes a lot of time, it is better to let it settle to the bottom, and only then filter.
So the next time you make it, select the right mixture (or use pure nitric acid, if there are nitrogen oxides, you can add a little urea to it, it
does not interfere with nitration), maintain a temperature of 10-15°C, gradually heat the mixture, and organize intensive mixing, especially at the
heating stage.
Well, the last photo is the finished product)) there is exactly 1 kg, but in two different jars. The red one contains fine-crystalline PETN (you can
see from the walls how much it stains the surfaces). in a transparent jar it is coarse-crystalline
   
[Edited on 9-9-2023 by DennyDevHE77]
|
|
|
Etanol
Hazard to Others
Posts: 162
Registered: 27-2-2012
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by DennyDevHE77 |
So, as I understand it you used the sulfur-nitrogen method of nitration in one stage, but you used the proportions for 70% nitric acid. You should
realize that too strong sulfur-nitrogen nitro mixtures oxidize PETN much easier than nitrates.
|
On the contrary, strong nitros mixtures usually form nitroesters very well.
I think it's not about the stiffness of the nitro mixture.
| Quote: Originally posted by ManyInterests |
I used 88g of PE (98%, it was technical grade),
I used 290ml of 90% HNO3 (purged of all NO2 contamination by blowing dry air through it) and 170ml of H2SO4 |
Have you checked the concentration of your sulfuric acid?
A salt-ice mixture is not required. A ice-water mixture is enough.
Do not hurry. Pentaritritole is poorly dissolved in HNO3-H2SO4 mix.
Add should take at least 15 minutes. After that, let the temperature very slowly rise to 20C. Wait 15 minutes. Never stop stirring!
Only if everything is fine, smoothly raise the temperature to 50C.
You may not heat the reaction mixture. Then, after stabilization in an aqueous solution of soda and recrystallization, the output of the product will
decrease by 5-10%.
[Edited on 9-9-2023 by Etanol]
|
|
|
DennyDevHE77
Hazard to Others
Posts: 162
Registered: 15-9-2014
Member Is Offline
Mood: No Mood
|
|
PETN appears to be a small exception to this pattern. Once I tried to nitrate it with a mixture of 95% nitric acid and 98% sulfuric acid, I almost got
a fountain, the temperature rose very sharply. A friend who used distilled nitric acid with phosphoric anhydride 99.7% with 98% sulfuric acid had a
problem and it ended in failure. Maybe for 90% the difference is not so significant, but the reaction proceeds much worse than with 70% and especially
58% nitric acid, perhaps due to a very thick mixture and poor heat removal. Therefore, in the presence of fuming nitric acid, the best option would be
to obtain PETN directly, without sulfuric acid.
And finally, I don’t remember a single method for producing PETN using highly concentrated nitro mixtures. In the books of Naum, Stettbacher and
Solonina for 1920-1930, PETN was obtained by esterification of pentaerythritol in >90% nitric acid, and straining with sulfuric acid (which now
sounds like a waste of sulfuric acid), there was a two-stage method, when sulfate was obtained and then nitrated > 90% nitric acid. Then they
switched to a one-stage process, without using sulfuric acid at all. And the method through dilute nitric acid and concentrated sulfuric acid seems to
have been invented by the Czechs in 2002, and it seems that this method was then spread to home hobbyists.
Well, it is obvious that for 90% of nitric acid and sulfuric acid a lot was taken, it is necessary to recalculate how much is needed according to the
schedule from Urbansky. But here I am no longer a helper((
It’s not for nothing that Ethanol mentioned this, nitration of pentaerythritol at too low a temperature 0-5°C leads to the formation of a certain
amount of PETRIN, this is pentaerythritol trinitrate, a liquid explosive at room temperature. It was previously used in the USA for all sorts of
plastisol explosives. On the other hand you can just add your pentaerythritol faster, it is important that the temperature is kept at 10-15°C
[Edited on 9-9-2023 by DennyDevHE77]
|
|
|
DennyDevHE77
Hazard to Others
Posts: 162
Registered: 15-9-2014
Member Is Offline
Mood: No Mood
|
|
Ethanol, since the administration will still transfer all the latest messages to another topic (and rightly so), let me ask, do you know how to
calculate the proportions from the diagram? This would be very useful to know.
I thought so, you need to select such a value of acid concentration so that it is on the lower border of region 1. After that, find out the final
concentration of acids, this point will be somewhere higher and to the left. Then you need to calculate how much nitric acid has decreased (or water
has been added) and create a proportion to this mixture. And here I’m already slowing down... Or maybe my logic is completely wrong.
If you know this, could you write down at your leisure how to count correctly. (Or maybe back in 2002 this was already covered here)
[Edited on 9-9-2023 by DennyDevHE77]
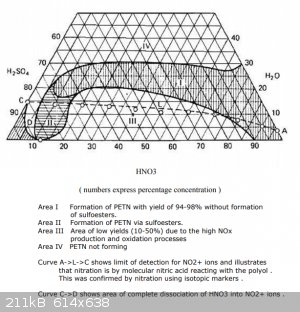
|
|
|
Etanol
Hazard to Others
Posts: 162
Registered: 27-2-2012
Member Is Offline
Mood: No Mood
|
|
You can’t count anything according to this diagram. It is not even known it refers to the initial acid or to the spent acid. There are no nitration
conditions, an excess of nitric acid to pentaritrite, no nitration time.
If you can find and show the source that Urbansky refers to, then we can do something.
There are reasons not to trust this diagram.
[Edited on 9-9-2023 by Etanol]
|
|
|
ManyInterests
National Hazard
Posts: 870
Registered: 19-5-2019
Member Is Offline
|
|
| Quote: | | Then the beaker with PETN is placed in a water bath with warm water. It is necessary to gradually raise its temperature to 25-30°C. The water in the
water bath is then replaced by very hot water and the main heating to 50°C is started. When the temperature of PETN reaches 50-52°C feel free to
take the beaker out of the water bath, and return it when the temperature drops below 50°C again. At this stage, the sulfo groups in PETN are
replaced by nitro groups, this is called the transesterification reaction. It starts at +35°C but reaches an acceptable rate at +50-60°C.
|
Ahh, so THAT'S why I got my yield so low. I guess if I am going to use the H2SO4 + HNO3 the HNO3 does need to be lower than RFNA. OK that does make
sense. If I am going to use nitric acid stronger than that, I should only use WFNA if/when I have it. It's starting to click.
| Quote: | And you really can not heat your PETN (but this will require longer cleaning), the yield will be on average 70-80%, or it is possible to heat for a
short time, as at 50°C for 7-10 min of heating the yield already becomes about 90%, at full transesterification the yield is 92%. I often produce
PETN, with 100g of pentaerythritol I consistently get ~220g.
In general, if you have 90%, you would be better off immediately nitrating with pure nitric acid, getting the same yield of 90%, but saving yourself
from lengthy cleaning. Just in case, I will point out that nitration is carried out with 90-99% nitric acid at the rate of 100 g of acid per 20 g of
pentaerythritol at a temperature of 20°C. This method is convenient for cleaning.
You see, PETN obtained by a two-stage or one-stage method through a nitro mixture is contaminated with mixed sulfoesters, which reduce its power and
stability, since previously PETN was produced in two stages industrially, this issue is generally well studied. So, especially if you did not heat the
mixture, you need to pour 9-10 parts of a 1% aqueous solution of sodium bicarbonate, and boil your PETN until it stops foaming. Instead of baking
soda, you can use, for example, magnesium oxide or sodium carbonate, but here it is better to keep the temperature at 90°C, which is less convenient;
at 100°C there will be slight decomposition. |
I'm still not sure what you mean by two-stage or one-stage. But I will take it at this point that I should do the following for my successive PETN
syntheses.
1: Either use 99% WFNA alone, or 70% HNO3 + H2SO4. If using WFNA and there is some yellowing, a touch of urea will clear it up.
2: don't let temperatures be too cold, keep at 10C and don't let it go above 15C until all the PE is added. I understand why. PETN needs that
temperature otherwise a trinitrate will form. This could also be why my yield was so bad in addition to other factors.
3: Let it stay at 25-30C for a while (30 minutes) and not necessarily heat it beyond that. But it can be heated to 40 or 50C for around 10 minutes.
4: After washing with lots of water and sodium carbonate water, boiling it in carbonate solution is a good idea? I assume a lot of stirring is needed
here.
I'll get to the recrystalization part later.
|
|
|
DennyDevHE77
Hazard to Others
Posts: 162
Registered: 15-9-2014
Member Is Offline
Mood: No Mood
|
|
Three methods of producing PETN are usually distinguished:
1. Production in one stage through nitric acid of 90-99% concentration (the main modern method of production of PETN in the industry).
2. Production in two stages. First, pentaerythritol sulfates are obtained by dissolving pentaerythritol in concentrated sulfuric acid. And then
nitrate the sulfates with fuming nitric acid at 50-60°C (obsolete method of PETN production in the industry).
3. Production in one stage through sulfuric-nitric acid mixture.
No, I repeat, the transesterification reaction, that is, the substitution of sulfogroups for nitro groups begins at +35°C, and goes more or less
quickly at 50-60°C. So after adding the last part of pentaerythritol to the nitro mixture, wait 5 min, and then you can immediately pour into water,
well or start heating.
The boiling water causes the PETN to float on its surface and itself agitates it on its surface. But a good idea from time to time is to sink down out
the PETN floating on the surface with a skimmer. Usually one hour of boiling is enough.
[Edited on 10-9-2023 by DennyDevHE77]
|
|
|
ManyInterests
National Hazard
Posts: 870
Registered: 19-5-2019
Member Is Offline
|
|
OK sounds simple. So upon the final addition, either dump or start heating. I'll probably start heating since I don't want any delay.
I'm taking a bit of a break from most chemistry for now. But I will be back at it in a month or so.
|
|
|
Etanol
Hazard to Others
Posts: 162
Registered: 27-2-2012
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by DennyDevHE77 |
The boiling water causes the PETN to float on its surface and itself agitates it on its surface. But a good idea from time to time is to sink down out
the PETN floating on the surface with a skimmer. Usually one hour of boiling is enough.
|
Boiling is an incorrect description of the procedure. Literally, stiring in a 1% aqueous solution of sodium bicarbonate at a temperature of about
90°C with pH control, not 100° C. Bicarbonate decomposes and hisses. It looks like a boil, but it is not a boil of water at 100C. When boiling at
100°C, the PETN decomposes partially.
| Quote: Originally posted by ManyInterests |
2: don't let temperatures be too cold, keep at 10C and don't let it go above 15C until all the PE is added. I understand why. PETN needs that
temperature otherwise a trinitrate will form. This could also be why my yield was so bad in addition to other factors.
|
The optimum temperature when adding pentaritrite is +5 ... +15°C.
There are two reactions that require exposure at elevated temperatures:
transformation of trinitrate into tetranitrate at 15...25°C.
and the transformation of sulfate into tetranitrate at 40...60°C.
These are two stages that require each control. You should not heat the mixture up to 50°C, while there are pentaeritritole and its trinitrate.
For WFNA 90%, you can increase its excess to 80 grams of WFNA to 10 grams of pentaeritritole and do without sulfuric acid and without heating,
That is, after adding, the mixture should be stirred 30...45 minutes at 20°C and pour into water with ice.
Or you can leave 50% excess of WFNA and thread by mixture:
10 g of pentaeritritole
30 g of WFNA 90%
20 g of sulfuric acid 96%
Temperature when adding PE is +5...+15°C,
then stirring at 15...20°C,
then stirring at 50...60°C,
then cool and pour into the water with ice.
Or
10 g of pentaeritritole
50 g of WFNA 90%
10 g of sulfuric acid 96%
Temperature when adding PE is +5...+15°C,
then stirring 30...45 min at 15...20°C,
without heating!
then cool and pour into the water with ice.
[Edited on 10-9-2023 by Etanol]
|
|
|
DennyDevHE77
Hazard to Others
Posts: 162
Registered: 15-9-2014
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Etanol | | Literally, stiring in a 1% aqueous solution of sodium bicarbonate at a temperature of about 90°C with pH control, not 100° C. Bicarbonate decomposes
and hisses. |
I've read about it in some industrial chemistry books. Usually they would heat the soda solution to 90°C, then add the PETN, and stir for an hour.
But I've also seen descriptions of laboratory methods where PETN was poured into the soda solution and heated to boiling, boiled until the foam
stopped forming.
Having tried both methods with baking soda and soda ash, I have come to the conclusion that when boiling with baking soda, a very small amount of PETN
is lost. In fact, I may have been losing them on the filter.
That said, when boiling with sodium carbonate (soda ash), the PETN decomposes significantly, and it's really worth maintaining 90°C there.
Also PETN boiled with magnesium oxide quite calmly, but then it was necessary to separate them in acetone.
In the photo below PETN is boiled at 105°C (tap water), here a little over 100 grams.
Thanks for that info, I was getting some trinitrate from nitration at 0-5°C. And so I thought (apparently wrongly) that at nitration at 10-15°C
trinitrate is no longer formed, but I always gradually heated PETN, so apparently without accidents. Then, in all likelihood, keeping PETN at 20-30°C
makes sense, I must apologize to OneEyedPyro. Another thing is how justified it is, will it help to raise the output by at least 5%?
In the near future I plan to get trinitrate, but unfortunately with high yield it can be obtained only through acetic anhydride, at nitration at low
temperature yield usually 40-50%. As far as I know, trinitrate can be treated with alcoholic alkali solution and bis-(nitratomethyl)oxetane can be
obtained. A fusible base for explosives. But I have not found much information on the forum.

|
|
|
Etanol
Hazard to Others
Posts: 162
Registered: 27-2-2012
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by DennyDevHE77 |
But I've also seen descriptions of laboratory methods where PETN was poured into the soda solution and heated to boiling, boiled until the foam
stopped forming.
|
I think this is a shorthand description.
Bicarbonate decomposes into carbonate when boiled.
This will help by 5-10% if all other conditions are met successfully, when the yield is 85 or 95%.
[Edited on 10-9-2023 by Etanol]
|
|
|
ManyInterests
National Hazard
Posts: 870
Registered: 19-5-2019
Member Is Offline
|
|
I am pushing the thread back up since I have a question to ask... two questions maybe.
The first one is since ammonium nitrate will work. I want to know if the nitrogen level of the ammonium nitrate matters all that much? Meaning if I
get my hands on cold-pack grade ammonium nitrate (27%) will it work, or does it have to 32%+ for it to work better? Ditto for aiding in RDX synth and
other chemical reactions.
The second question: Since I ordered a good deal of pure calcium nitrate, and that can be used to make any other nitrate I want, if I do a double
displacement reaction with ammonium carbonate/bicarbonate, will the resulting ammonium nitrate be 34% or will it be a lower nitrate content? If it is
a lower content, is it possible to increase the nitrogen content of that AN? What about using the sodium bisulfate + ammonia method?
I am saying this because what I thought might be ammonium nitrate from the fertilizer section of my hardware store proved to be a disappointment.
|
|
|
Microtek
National Hazard
Posts: 845
Registered: 23-9-2002
Member Is Offline
Mood: No Mood
|
|
Pure ammonium nitrate is 35% nitrogen. If you get something with less than that, it has some impurities. If you don't know what these impurities are,
you are shooting in the dark if you use it in nitrations. Some cold packs contain urea instead of AN but this is not the issue here, since urea has a
higher nitrogen content than AN. It could conceivably be water or maybe ammonium chloride, but you will have to do some actual chemical analysis if
you can't find a proper list of ingredients.
Your second question makes me think you should read up on introductory inorganic chemistry. I suggest finding some high school texts as this will
answer all of these questions. It might also save you some money in the future.
|
|
|
ManyInterests
National Hazard
Posts: 870
Registered: 19-5-2019
Member Is Offline
|
|
Bumping this up since I saw a video on YouTube that described a one-pot process. While it is interesting (and not very complicated. It simply involved
a larger amount of some reagents and a steady increase of heat to a point. Similar to picric acid actually). But he did mention in purifying the final
product, he said that either sodium metabisulfite (which I have) or 91% isopropyl alcohol will work to purify it. Is this true? Because I can get any
91% or 99% IPA much more cheaply and easily than sodium metabisulfite.
|
|
|
Sir_Gawain
Hazard to Others
Posts: 317
Registered: 12-10-2022
Location: Due South of Due West
Member Is Offline
Mood: Way less sad
|
|
Was it this one?
“Alchemy is trying to turn things yellow; chemistry is trying to avoid things turning yellow.” -Tom deP.
|
|
|
ManyInterests
National Hazard
Posts: 870
Registered: 19-5-2019
Member Is Offline
|
|
Nope that is the older one (still excellent and I intend to follow it as well to see which one gets the best yield), but I am referring to this one:
https://youtu.be/H0OI3hyjkFM
edit: He starts to talk about recrystalization and final purification around the 23:35 mark.
Edit2: While I'm at this. While I am looking up on the red water issue and how to properly dispose of it. Is there some way of safely neutralizing it
so it can be poured down the drain, or does it have to be incinerated? If that is the case, will a blowtorch do? Anything I should know about burning
this stuff from a safety perspective? Beyond the fumes and such?
[Edited on 14-12-2023 by ManyInterests]
[Edited on 14-12-2023 by ManyInterests]
|
|
|
Sir_Gawain
Hazard to Others
Posts: 317
Registered: 12-10-2022
Location: Due South of Due West
Member Is Offline
Mood: Way less sad
|
|
Just evaporate it down and throw it on a fire.
“Alchemy is trying to turn things yellow; chemistry is trying to avoid things turning yellow.” -Tom deP.
|
|
|
ManyInterests
National Hazard
Posts: 870
Registered: 19-5-2019
Member Is Offline
|
|
You see that's my issue. I always overcomplicate everything. That sound very simple. Finding a place to safely incinerate it will be a mild challenge
(I live in a big city. I can't exactly go to a public park with people around and burn stuff) but it is dooable.
|
|
|
OneEyedPyro
Hazard to Others
Posts: 280
Registered: 7-10-2015
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by ManyInterests |
Edit2: While I'm at this. While I am looking up on the red water issue and how to properly dispose of it. Is there some way of safely neutralizing it
so it can be poured down the drain, or does it have to be incinerated? If that is the case, will a blowtorch do? Anything I should know about burning
this stuff from a safety perspective? Beyond the fumes and such?
[Edited on 14-12-2023 by ManyInterests]
[Edited on 14-12-2023 by ManyInterests] |
There's a time and place for everything. Can't you simply store it until a good opportunity arises to burn it? There's no real eco friendly way to get
rid of the byproducts I'm aware of but incineration is probably your best bet. Maybe prepare a campfire as in ready to light ahead of time then come
back at night with the waste, light the fire and walk away.
This is assuming it's not a fire hazard area. I'd evaporate it down in a disposable aluminum cake pan and place it on the fire.
|
|
|
chem101st
Harmless
Posts: 8
Registered: 1-12-2023
Member Is Offline
Mood: No Mood
|
|
Dugan uses an overhead stirrer in that video. Is that necessary due to viscosity or something? Or is a standard stir bar fine?
|
|
|
ManyInterests
National Hazard
Posts: 870
Registered: 19-5-2019
Member Is Offline
|
|
| Quote: Originally posted by OneEyedPyro | | Quote: Originally posted by ManyInterests |
Edit2: While I'm at this. While I am looking up on the red water issue and how to properly dispose of it. Is there some way of safely neutralizing it
so it can be poured down the drain, or does it have to be incinerated? If that is the case, will a blowtorch do? Anything I should know about burning
this stuff from a safety perspective? Beyond the fumes and such?
[Edited on 14-12-2023 by ManyInterests]
[Edited on 14-12-2023 by ManyInterests] |
There's a time and place for everything. Can't you simply store it until a good opportunity arises to burn it? There's no real eco friendly way to get
rid of the byproducts I'm aware of but incineration is probably your best bet. Maybe prepare a campfire as in ready to light ahead of time then come
back at night with the waste, light the fire and walk away.
This is assuming it's not a fire hazard area. I'd evaporate it down in a disposable aluminum cake pan and place it on the fire.
|
No I can store it. I've store waste products until I could safely dispose of them for a while. I know how and where to do that. If incinerating it is
the only option, then that is what I will do. I will find an isolated place in a park where you can start a small camp or cook fire, but I can also go
into the woods when fire alerts are at a minimum (this is usually during winter time or after it has rained, and the ground is quite wet and moist...)
and get my fire extinguisher and other fire fighting gear to make sure no issues arise just in case.
| Quote: Originally posted by chem101st | | Dugan uses an overhead stirrer in that video. Is that necessary due to viscosity or something? Or is a standard stir bar fine? |
There's another video where someone just uses a stirbar. Dugan's method is only a little different than the other guy (who uses a 2-step method of
making TNT).
However I do have one question: Dugan used isopropyl alcohol in place of sodium sulfite/bisulfite to purify it. Will 99% isopropyl alcohol work
because TNT does have some slight solubility in water I would rather use something it isn't soluble in to clean it... unless I am mistaken. I know
that TNT is soluble in ethanol more than water, but what about IPA?
|
|
|
Sir_Gawain
Hazard to Others
Posts: 317
Registered: 12-10-2022
Location: Due South of Due West
Member Is Offline
Mood: Way less sad
|
|
Trinitrotoluene is much more soluble in isopropyl alcohol than water. Didn’t Dugan recrystallize it from IPA?
“Alchemy is trying to turn things yellow; chemistry is trying to avoid things turning yellow.” -Tom deP.
|
|
|
DennyDevHE77
Hazard to Others
Posts: 162
Registered: 15-9-2014
Member Is Offline
Mood: No Mood
|
|
The big advantage here is that cleaning is optional. Of course, it can be recrystallized from alcohol or acetone, it can be melted in boiling water
and mixed with compressed air, or it can be done with the addition of sodium sulfite, this is generally an industrial cleaning method, it can be
compressed in layers of newspapers so that they absorb dinitro derivatives. You can add calcium silicate so that it absorbs TNT oil.
|
|
|
ManyInterests
National Hazard
Posts: 870
Registered: 19-5-2019
Member Is Offline
|
|
| Quote: Originally posted by DennyDevHE77 | | The big advantage here is that cleaning is optional. Of course, it can be recrystallized from alcohol or acetone, it can be melted in boiling water
and mixed with compressed air, or it can be done with the addition of sodium sulfite, this is generally an industrial cleaning method, it can be
compressed in layers of newspapers so that they absorb dinitro derivatives. You can add calcium silicate so that it absorbs TNT oil.
|
Actually way, I looked again into the solubility. It is practically soluble in water. Only 0.13g per 1000ml of 20C water. Yeah washing it with cold
water is better.
I'll use sodium metabisulfite to clean it. It turns into bisulfite when it is put in water..
While it can be 'optional' to clean it, for me it is part of the excitement of making such material is to also make it as close as possible to the
final industrial product. I do it as much as a point of pride as it is from a practical standpoint.
It is like how I did everything I could to purify my picric acid. Even though I realized that using salicylic acid yields such a purer product right
off the bat that the only thing needed is to wash away excess sulfuric acid and it is good as is. As well as drying it and changing paper towels
frequently since I believe that those towels loved to suck up the acid as well.
|
|
|
| Pages:
1
..
10
11
12
13 |









