| Pages:
1
2
3
4 |
chemoleo
Biochemicus Energeticus

Posts: 3005
Registered: 23-7-2003
Location: England Germany
Member Is Offline
Mood: crystalline
|
|
Please remind me, wasn't there a thread recently with excellent pictures on some manganese oxide derivative, which was used for oxidative purposes?
I'll happily delete this post once it's clarified.
Never Stop to Begin, and Never Begin to Stop...
Tolerance is good. But not with the intolerant! (Wilhelm Busch)
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
| Quote: | Originally posted by chemoleo
Please remind me, wasn't there a thread recently with excellent pictures on some manganese oxide derivative, which was used for oxidative purposes?
I'll happily delete this post once it's clarified. |
Yeah , the other manganic alum reducing reagent thread is
https://sciencemadness.org/talk/viewthread.php?tid=6882#pid7...
maybe even the threads should merge ?
|
|
|
chemoleo
Biochemicus Energeticus
Posts: 3005
Registered: 23-7-2003
Location: England Germany
Member Is Offline
Mood: crystalline
|
|
Hilski, thanks first of all for your excellent contributions.
I merged this thread with the previous one on this matter.
Please send me a U2U if you aren't happy with the title.
I'd like to keep similar threads together because it will make searching for it so much easier, and it'll keep the forum more condensed.
PS Perhaps the info/pics on ammonium manganic alum should be written together for the pre-puplication forum?
Never Stop to Begin, and Never Begin to Stop...
Tolerance is good. But not with the intolerant! (Wilhelm Busch)
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Just a bit of additional information which could be relevant and useful in strategies for original preparation of a manganic alum oxidation reagent ,
for adjustment / correction of the initial oxidation state , as well as an alternative chemical means for regeneration of
the spent reagent .
Potassium permanganate can be used to oxidize the
other manganous compounds present to the manganic
form . Of course by using additional manganate in the
form of permanganate , your quantity of regenerated
reagent will grow and accumulate . But this would provide a quick means of regeneration , in the case where time may be an issue and it is
inconvenient to wait for perhaps days for an electrolytic regeneration to complete .
The proportions would have to be worked out , as applies
to the manganous alums regeneration , and the additional materials of ammonium or potassium donors needed to maintain the alum composition for the
reagent ,
producible by such a chemical regeneration via permanganate .
Attached is a patent which shows how this proceeds
in the preparation / regeneration of a similar manganic
oxidation reagent . This may be of interest and adaptable to the alums as well .
Attachment: US837777 Usefulness of Permanganate for regenerating Manganous to Manganic reagent.pdf (236kB)
This file has been downloaded 1220 times
|
|
|
Hilski
Hazard to Others
Posts: 197
Registered: 13-9-2006
Member Is Offline
Mood: No Mood
|
|
Thanks for that post, Roscoe.
I had been thinking about ways one could go about regenerating the oxidizer in a more expedient manner. KMNO4 had came to mind, but I have no idea
how to go about calculating proper proportions. I also wondered how adding another element (K) would effect the behavior of the oxidizer both during
the reaction with toluene, and during electrolytic regeneration.
Perhaps a small specific amount of the permanganate could be used in addition to the electrolytic process to say, cut regeneration times in half. I
also wonder if something like ammonium or potassium persulfate could be used to achieve the same results?
I guess seeing as how I already have about 2 liters of the alum prepared and fully functional, that I should be the one answering these questions
instead of asking them. But what fun is that? 
\"They that can give up essential liberty
to obtain a little temporary safety
deserve neither liberty nor safety. \"
- Benjamin Franklin
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
I have wondered if even hydrogen peroxide , or
perhaps acetone peroxide , or MEKP could be used for regeneration of the manganous alum to the manganic alum . It sure would save hours of
electrolysis to be able to stir in the stuff and boil off the inert byproduct .
Yeah I would think that the persulfates should also
work the same way with their respective potassium
or ammonium alums . You would have to keep an
inventory of your reaction mixture content with the
desired proportions of the respective ions , and
work out the amounts of what you add during regeneration , so that you keep the composition
of the reagent consistent with its proper stoichiometric proportions for the desired alum formation .
I haven't done the math , so I am not sure how the
numbers crunch on this idea ....it just looked like an
interesting possibility , particularly on the initial formulation of the potassium alum ....it looked like
that making the reagent in the manganic form ready
for use without electrolyzing for the first run , could
be done by forming some of the potassium sulfate and
manganic sulfate content from potassium permanganate
plus H2SO4 , eliminating or reducing the need for the
electrolysis . I think there is even some surplus oxidizing
capability there for oxidizing the bulk manganous sulfate
to the manganic state . So it appeared to be a way of shortcutting the workup for the initial alum ....if nothing else . What would be the economy
of this as compared
with just using electrolysis alone on the manganous sulfate alum is another matter which could make it not worthwhile .
BTW I found an analogous process is possible for chromium also , as is used here for manganese compounds ....but of course the chromium reagent
would be a way more expensive method .
I am still inclined to believe that the ferrous salt and
its alums would also work for this process , and also
other polyvalent metal salts .....although my interest
would be focused on the cheap ones like manganese
and iron
|
|
|
Organikum
resurrected
Posts: 2330
Registered: 12-10-2002
Location: Europe
Member Is Offline
Mood: frustrated
|
|
Vanadium is told to work with H2O2 even in situ with no need for transferring something from one cell to another. The article should be in the toluene
to b-dehyde thread.
/ORG
|
|
|
Hilski
Hazard to Others
Posts: 197
Registered: 13-9-2006
Member Is Offline
Mood: No Mood
|
|
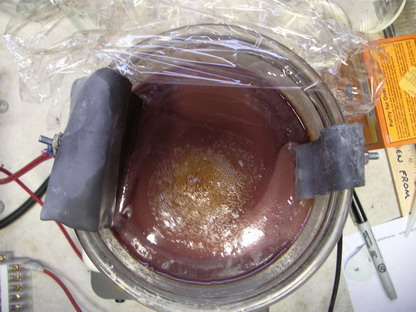
I guess all in all, passing current through a non-divided cell for 24 hours isn't really all that bad. Unless one needs to regenerate the oxidizer
like RIGHT NOW, then there just isn't a more economical way to do so. Here are a couple of pictures of the electrolytic cell regenerating the MAA
recently. 4 amps have been passing through the cell for about 10-12 hours in the photos. I think it was still needing about 10-12 more to be fully
regenerated. You can see the large quantity of yellow precipitate (MAS) still sitting at the bottom of the jar. This is because I have crammed so
much oxidizer into this jar with not enough acid that the mag stirrer has a hard time keeping it all in suspension. A little poking around with a
glass rod gets everything mixed up again though, so it really isn't a problem. If you look at the second picture, you can see a brown substance
floating on top, in the center of the jar. This is mostly benzoic acid floating on top of the saturated acid solution. It is due to left over
toluene and/or benzaldehyde from a previous reaction. It's hard to get every last little bit of organic material out of the mix, but it really
doesn't affect anything. It just smells REALLY strong once too much builds up in the cell.

| Quote: | Vanadium is told to work with H2O2 even in situ with no need for transferring something from one cell to another. The article should be in the toluene
to b-dehyde thread.
/ORG
|
I recall seeing something about venadium sulfate (I think) being used for this purpose with H2O2. Not having to transfer anything back and forth is
always a plus. That's one reason I like this MAA oxidizer so much. Once the oxidation is complete, and the aldehyde has been extracted, just drop in
two electrodes and turn on the PSU. No membrane, or other special apparatus required.
[Edited on 26-1-2007 by Hilski]
\"They that can give up essential liberty
to obtain a little temporary safety
deserve neither liberty nor safety. \"
- Benjamin Franklin
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
@Organikum
As I think I understand this process so far .....
That patent which I posted above , US837777 , is directly related .....but superior to
the " neograviton method " which you posted early on the Benzaldehyde from Toluene thread .
The earlier patent US780404 , by the same inventor Max Bazlen , is a patent which you listed among others in the other thread . The yield of
benzaldehyde from toluene by oxidation with manganese (IV) sulfate , manganese persulfate , is reported as nearly quantitative , which generally means
>95% , with no byproduct benzoic acid
from any overoxidation .
The subsequent patent US837777 simply advances the
same process using Potassium Permanganate for generating the dark brown colored
Manganese ( IV ) "per"sulfate , Mg(SO4)2 .
The (IV) valency variant is a yet still more active oxidizer than the also useful *green* colored Manganese ( III ) analogue , or Mangan " ic "
Sulfate , Mg2(SO4)3 .
Reduction of the (IV) and/or the (III) compound during oxidation of toluene to benzaldehyde produces
the fully reduced Manganese ( II ) Mangan " ous " Sulfate , MgSO4 as the spent reagent , which is regenerated ,
by electrolytic oxidation , stepped back up to its (III) and
further to its original (IV) oxidation state for reuse .
CRC gives a range of colorations from pink through red for the ( II ) manganous sulfates depending upon the level of hydration , including the alum
with ammonium sulfate , which BTW is a hexahydrate .
Oxidation to the ( III ) state is described as producing a
green color for the pure manganese(III) or mangan" ic "
sulfate .
I am thinking that the mixing of the green in with the incompletely regenerated red is what gives the darkening towards brown .....it could even pass
through a tar black color first and then lighten slightly to more distinct brown for the final transition to the brown coloration of the manganese (
IV ) sulfate , manganese persulfate . IIRC the persulfate is only quasi-stable and only in a range of fairly concentrated H2SO4 .
Anyway I suppose the advantage realized for any
"hyper-regeneration" being applied chemically using permanganate , or perhaps peroxide if possible , or by electrolysis of the manganic sulfate to
the even higher (IV) persulfate is that an additional amount of toluene may be converted to benzaldehyde by the same quantity of Manganese ion present
, if its oxidation state is at maximum , which is the persulfate .
I haven't gone back and checked the details on the
regenerations by electrolysis which have been pictured
and described , to see if anyone allowed the regeneration
to keep running longer than required to produce the pink
or red material , even where the alum double salt is being
used ......to see if it will continue to oxidize further to the
solid brown color as would indicate the persulfate .
Edit: I just went back and reviewed this thread , and it looks like the ammonium alum manganese sulfate follows the US808095 patent description
nicely , with regards to color
changes , and other details . Whether or not the yield or
the volume efficiency for this method could be improved by running the regeneration electrolysis longer , might be a worthwhile experiment . It might
be necessary to have some
additional amount of manganese sulfate present which is
beyond that amount which will form the alum in combination with the ammonium sulfate present . The alum itself may be limiting with respect to
formation of the persulfate .....and
of course the acid concentration would have to be within the allowable range also in order to permit its formation from
any added time of electrolysis . Color change and gas evolution from the anode would tell the tale on whether
this effort is worthwhile , along with of course comparison of the yield and how much toluene may be reacted with a given quantity of the regenerated
oxidation reagent .
But the typical yield for the alum Mn(III) process is 80% , compared with >95% reported for the non-alum Mn(IV)
process , which should also have a better volume yield ,
although it would depend on the volume of acid ( I haven't checked this to be sure ) . So it would seem that using the
Bazlen patent method based on the Mn (IV) sulfate would be superior .
Quantitative yield is as good as it gets
[Edited on 26-1-2007 by Rosco Bodine]
|
|
|
Hilski
Hazard to Others
Posts: 197
Registered: 13-9-2006
Member Is Offline
Mood: No Mood
|
|
| Quote: | to see if anyone allowed the regeneration
to keep running longer than required to produce the pink
or red material , even where the alum double salt is being
used ......to see if it will continue to oxidize further to the
solid brown color as would indicate the persulfate . |
I have let it go longer than required, and a brownish solution does result, although I didn't know why the brown color was present. I haven't
distilled the the aldehyde from the reaction with the brown(ish) oxidizer yet, so I don't know how it might have affected yields.
\"They that can give up essential liberty
to obtain a little temporary safety
deserve neither liberty nor safety. \"
- Benjamin Franklin
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
The brown material is precisely what you want for maximum yield . And you can use more toluene
also .....I haven't done the math , but offhand
maybe 50% more toluene per reaction with the same amount of " hyper" regenerated brown reagent ....
I'll have to check the math on this to be certain .
Attached is the original Bazlen patent US780404
concerning near quantitative yield of benzaldehyde from toluene via oxidation by manganese (IV) sulfate ,
manganese persulfate , which may be regenerated
by electrolysis .
[Edited on 27-1-2007 by Rosco Bodine]
Attachment: US780404 Manganic(IV) Sulfate electrolysis regenerated.pdf (173kB)
This file has been downloaded 1447 times
|
|
|
Hilski
Hazard to Others
Posts: 197
Registered: 13-9-2006
Member Is Offline
Mood: No Mood
|
|
Some experiments
As I mentioned in another thread, I would like to set up a functional electrolytic cell that would produce Manganese(IV) sulfate aka (MnSO4)2 from
MnSO4 in 55% H2SO4. I loosely followed the guidelines from This Patent to know what reagents/ratios etc to use. Also, one of my goals was to find out whether Tyvek (a commonly available material
manufactured by Dupont) would make a sufficient membrane for a divided electrolytic cell. The following are some details and a few pictures of what
I've come up with so far.
Chemicals and materials:
Anolyte:
MnSO4 - 50 grams disolved in 504 grams H2SO4 (55%)
Catholyte:
55% H2SO4
Cell construction:
Cathode compartment: A small HDPE container with holes drilled in the sides. Inside the HDPE container is placed a Tyvek bag which acts as a
microporous membrane to allow for ion echange. The cathode is 0.5 inches wide X 6 inches long and is made of lead sheet. The compartment is filled
with 55% H2SO4 and placed inside the glass beaker which will serve as the anode compartment.
Anode compartment: A 600ml Kimax beaker was used as the anode compartment. A lead anode, 1 inch wide X 6 inches long was placed in the compartment
along with a solution of 50 grams MnSO4 dissolved in 504 grams 55%H2SO4. This compartment is where (hopefully) a solution of Manganese(IV) sulfate
will be produced electrolytically.
The empty cell:

The cell after the pink MnSO4/H2SO4 solution has been added:

I hooked up the computer power supply I used for this project, and set the voltage to 5 volts. I chose this voltage because it's what I had used in
the past to regenerate the manganous ammonium alum, and it has worked well. I switched on the power, and a dark red color, similar to that of the
aforementioned alum, poured off of the anode.


I didn't use any kind of magnetic or mechanical stirring for this project, but if I decide to scale it up, I will definately add that to make the
process a lot faster and more efficient. For this small cell I just used a glass rod to stir things up occasionally.
Here is the cell after about 10 minutes:

And again at about 1 hour:

After about an hour and a half, the solution in the cell had turned a very dark, opaque burgundy color and didn't appear to be making any further
progress. I checked the current and saw that it was only pulling about 1.5 amps, and was generating almost no heat at all. So at that point I
decided to switch from 5 volts to 12 to see if that made a difference. Immediately upon switching on the 12 volt power, a brown color was generated
at the anode, along with more vigorous oxygen production. The hydrogen evolution at the cathode was enough that it started splashing the catholyte
out of the compartment. I also noticed the the cell started to heat up a good bit (as expected) and the current draw shot up to over 6 amps.
Note the brown color starting to form around the anode

At that point, I decided to place a 50 watt DC lightbulb in series with the cell to help control the current. As of now, the cell is pulling about 3
amps, and gas evolution is not nearly as bad as it was before. There doesn't seem to be nearly as much heat produced as before either.

The cell has been running for a couple of hours now, and as soon as there has been significant change (or lack thereof) I will update the post with
further results. At this point, I am optimistic that Tyvek does indeed make for a suitable membrane for divided electrolytic cells.
UPDATE:
At about the 3 hour mark I went to check on the cell and found what appears to be a successful experiment. Even with no stirring, the contents of the
anode compartment were a deep brown, almost black color. The heat in the cell was a lot lower than I expected, less than 40C for sure. Now I just
need to figure out exactly how long to this thing go to get 100% conversion to the persulfate.



Here are some links to high res copies of the last 3 images. The small ones are a little hard to see.
http://www.sciencemadness.org/scipics//Hilski/persulfate_cel...
http://www.sciencemadness.org/scipics//Hilski/persulfate_cel...
http://www.sciencemadness.org/scipics//Hilski/persulfate_cel...
Questions, comments or suggestions about any of this are welcome. Ideas for scaling up would be great as well.
-Hilski
[Edited on 1-2-2007 by Hilski]
\"They that can give up essential liberty
to obtain a little temporary safety
deserve neither liberty nor safety. \"
- Benjamin Franklin
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
That flouropolymer braid fly line would probably be good for lacing the tyvek snugly around the top of a wide mouth bottle ....but there is another
idea that may be better .
Get one of those clear polyethylene food storage cylinders that has the snap on poly lid and test it to
see if you put a sheet of tyvek across the top and
depress some slack in the middle of it , if the lid
will still snap down into place , sandwiching the
tyvek in the seal groove in the lid and securing it
in place . The tyvek will probably wrinkle a bit ,
but if the sealing groove in the lid will seat over the
tyvek , and secure it in place .....then you can use
the lid as a retaining ring to secure the tyvek liner
bag to the rim of the container . You can take a
razor knife or boxcutter and cut a big hole in the
lid , but leave the outside perimeter intact for use
as a retaining ring . Some dots of hotmelt glue might work for securing the tyvek to the inside of the storage cylinder . Leave slack in the tyvek
where it loops
over the sealing edge , so that when you seat the ring it doesn't pull the liner taught and pop it loose . If you
make the lead cathode cylindrical , maybe 1/4 to 1/3
the diameter of the inside of the catholyte compartment would be about right , bubbles will have room to escape
without the foam climbing the inside of the tyvek . A plastic trivet like they use in the bottom of those lettuce keepers would make a good bottom
spacer , or just use
another smaller diameter plastic lid on which to rest the bottom of the cathode , bore holes in it too if you like .
Heavy solid copper wire could probably be used to make
a tripod suspension bridle for the cathode and catholyte
compartment , hanging it from a suspension harness as if it were an ornamental hanging basket planter . I would
form a ring of the solid wire all the way around the plastic
rim of the container underneath that stiffening ridge and
make attachment loops to that wire ring , rather than
just going through small holes made in the plastic .....
because that plastic softens and weakens when it gets hot , and the attachment points might stretch and tear loose .
You could do a similar thing with some polyethylene
braided cord if you can find some small enough diameter
to work with .
Another strategy is heat welding .....tyvek could stick very
well to the rim of a polyethylene container which was gently
melted at the edge , just to the point of inflammation then quickly blown out like a candle , the tyvek pressed quickly
and firmly against the melt as it solidifies . Or just the hot
tip of a soldering iron might be used to " spot weld " the
tyvek at points like a dotted line , to " stitch " it to the
rim of the container . Poking holes through with a hot
nail and actually stitching it with a sailmakers needle
and the fluropolymer line is another option for lashing it down
to the rim of the cathode compartment . Slippery stuff to
handle , but there's a few ways of managing it .
[Edited on 1-2-2007 by Rosco Bodine]
|
|
|
Hilski
Hazard to Others
Posts: 197
Registered: 13-9-2006
Member Is Offline
Mood: No Mood
|
|
Those are all good ideas, and the type of thing I would need to address if scaling up. But one thing I would like to try is eliminating the plastic
cathode container altogether. I want to make sort of a sleeve to put the cathode in that is tall enough to cover most the cathode and that has enough
volume to hold enough catholyte to keep the cathode mostly covered all the time. Then, after filling the pouch with the 55% H2SO4 solution, I would
use a zip tie to secure the top of the pouch around the top of the cathode to attach the tyvek in place directly to the cathode.
If I do end up using a plastic container, another thing I had though about was to use one of those tall, flat HDPE bottles that hand lotions and
shower gel etc come in. The bottles are tall enough to be able line with tyvek and fit a large cathode in. But they are flat enough not to take up
too much space when stood upright inside a larger glass or plastic container, in an arrangement similar to what I used in my small scale experiment.
\"They that can give up essential liberty
to obtain a little temporary safety
deserve neither liberty nor safety. \"
- Benjamin Franklin
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
I definitely believe you should have some sort of rigid
compartment to keep everything positioned coaxially ,
so that hydrogen gas evolution from the cathode can
be managed smoothly , without " gurgling " and splashing .
Those red plastic press on cover " coffee cans " that
13 ounces of coffee is sold , are HDPE , 4" diameter
and about 5" tall might make a good compartment .
Something smaller in diameter and taller form cylinder
would be like a shampoo bottle . But the more narrow
and tall the container , the more of a geyser of liquid
will be carried up by the hydrogen bubbles , so your choice is going to set a limit on the current you can
manage .
|
|
|
Hilski
Hazard to Others
Posts: 197
Registered: 13-9-2006
Member Is Offline
Mood: No Mood
|
|
| Quote: | Those red plastic press on cover " coffee cans " that
13 ounces of coffee is sold , are HDPE , 4" diameter
and about 5" tall might make a good compartment . |
My thoughts exactly. I just salvaged one of those from the pantry. You are right about the bubbling and foaming being worse with a narrower
container, but I want to be able to accomplish effective stirring within the anode compartment, which the cathode compartment will be sitting in. So
unless I use a pretty large container for the anode compartment (like an 8 or 12 liter plastic bucket) then I need to keep the cathode compartment as
small as I can. Using a plastic bucket for this would be great, except for the fact that I have no way of heating the contents of said bucket to 50C
for the actual toluene oxidation. I could transfer the persulfate solution to the 2 liter glass jar like I use for the alum, and heat it the same
way, but it would be nice not to have to transfer the solution around if I don't need to.
Thanks for the suggestions.
Hilski
\"They that can give up essential liberty
to obtain a little temporary safety
deserve neither liberty nor safety. \"
- Benjamin Franklin
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
It would probably be better to put the tyvek around the *outside* of the perforated plastic form , as opposed to
having it inside like a liner bag . I just think that using
the rigid compartment as an inner form around which the
tyvek is stretched .....would simplify things greatly all around .....instead of " all inside "
This would definitely be a better arrangement with the smaller diameter cathode compartments .
Since this is the cathode also , a carbon rod or a piece of
copper or stainless rod or pipe might work fine , as no worries about oxidation and the hydrogen blanket on the surface should provide a lot of "
anti-erosion " by " cathodic protection " .
[Edited on 1-2-2007 by Rosco Bodine]
|
|
|
Eclectic
National Hazard
Posts: 899
Registered: 14-11-2004
Member Is Offline
Mood: Obsessive
|
|
Maybe one of these would work:

http://www.maycocolors.com/shapes/shapeDetails.cfm?shape=101...
Low fired porous ceramic bisque.
|
|
|
moxsnixs
Harmless
Posts: 6
Registered: 9-2-2007
Member Is Offline
Mood: No Mood
|
|
In cycloknights original post he reacted MnO2 with HCL then with H2SO4 to get MnSO4. Anyone have a more detailed procedure?
|
|
|
jimmyboy
Hazard to Others
Posts: 235
Registered: 1-3-2004
Location: Texas
Member Is Offline
Mood: No Mood
|
|
So overall - what is the best method - opinions? toluene/persulfate or reflux/oxidation of cinnamaldehyde - this is abit more involved but not too
hard - some manganese - electrodes - power supply - it looks like you really need that stirbar - actually the supplies are easier to get - you have to
look a little for cinnamon oil - i am just looking at it from a yield standpoint
|
|
|
Hilski
Hazard to Others
Posts: 197
Registered: 13-9-2006
Member Is Offline
Mood: No Mood
|
|
Unless huge quantities are needed, oxidation of toluene by manganese compounds would be the simplest, cheapest and most OTC in my opinion. I have
never tried the cinnamon oil route, due to the fact that the stuff is actually more expensive than just buying benzaldehyde. A one gallon container
will easily hold enough oxidizer/H2SO4 to produce 200g or more of benzaldehyde or benzoic acid, formaldehyde, formic acid, acetaldehyde (maybe) etc.
| Quote: | | In cycloknights original post he reacted MnO2 with HCL then with H2SO4 to get MnSO4. Anyone have a more detailed procedure? |
Although making the MnSO4 from MnO2 isn't hard, it is much better to just order some fertilizer grade MnSO4 from the net. (NH4)SO4 can be found
cheaply and in large quantities in any decent garden center or nursery supply type store.
Here is a thread that goes into detail on some different methods for making MnSO4
https://sciencemadness.org/talk/viewthread.php?tid=6777#pid7...
[Edited on 10-2-2007 by Hilski]
\"They that can give up essential liberty
to obtain a little temporary safety
deserve neither liberty nor safety. \"
- Benjamin Franklin
|
|
|
moxsnixs
Harmless
Posts: 6
Registered: 9-2-2007
Member Is Offline
Mood: No Mood
|
|
OK thanks I added the Na2CO3 and got the MnCO3 but the final step with H2SO4 wasnt really hit on. Do you add it to the MnCO3 or filter the MnCO3 and
add it to that?
I would buy it but not in the USA and to buy it here requires a 3hr drive one way, get a hotel room then try to find where its sold.So making it
would save alot of BS .Thanks
|
|
|
not_important
International Hazard
Posts: 3873
Registered: 21-7-2006
Member Is Offline
Mood: No Mood
|
|
| Quote: | Originally posted by moxsnixs
OK thanks I added the Na2CO3 and got the MnCO3 but the final step with H2SO4 wasnt really hit on. Do you add it to the MnCO3 or filter the MnCO3 and
add it to that?
I would buy it but not in the USA and to buy it here requires a 3hr drive one way, get a hotel room then try to find where its sold.So making it
would save alot of BS .Thanks |
Wash the MnCO3by to get rid of all the sodium salts, cover with water, and slowly add H2SO4 with stirring. Might be best to pre-dilution the sulfuric
acid if its concentrated. You want enough water to dissolve the MnSO4 formed.
If you don't protect the wet MnCO3 from air it will be oxidised and turn dark. Not a big deal if you're make the Mn(3+)-alum from it.
If your MnO2 wasn't pure, or if your HCl has iron in it, it might be best to set aside about a tenth of the MnCO3. Add just enough acid to the
remaining 90% to get it into solution, you'll not want an excess. Bring it to a gentle boil to drive off CO2, cool to it's just warm, add the
remaining MnCO3 with stirring, continue to stir and bring to a boil for a minute or two. Then let it cool, filter off the precipitate which is MnCO3,
iron carbonates/hydroxides, and hydrated oxides of manganese in 3+ and 4+ oxidation state.
|
|
|
moxsnixs
Harmless
Posts: 6
Registered: 9-2-2007
Member Is Offline
Mood: No Mood
|
|
And then evap the MnSO4 solution to obtain the powder correct? because I did and got a faitly pink\white powder
|
|
|
Hilski
Hazard to Others
Posts: 197
Registered: 13-9-2006
Member Is Offline
Mood: No Mood
|
|
| Quote: | | And then evap the MnSO4 solution to obtain the powder correct? because I did and got a faitly pink\white powder |
Correct.
Just FYI MnSO4 can be found on ebay. I think I remember seeing it sold for use on palm trees or something similar.
\"They that can give up essential liberty
to obtain a little temporary safety
deserve neither liberty nor safety. \"
- Benjamin Franklin
|
|
|
| Pages:
1
2
3
4 |









{kind=link}
{kind=link}
{kind=link}