guy
National Hazard

Posts: 982
Registered: 14-4-2004
Location: California, USA
Member Is Offline
Mood: Catalytic!
|
|
Chloranil
I know the standard way of making chloranil is by mixing NaClO3/HCl with phenol. But I do readily have access to chlorates, I wonder if it is
possilble to use TCCA or NaDCCA? I don't see how it wouldn't work but I just want to make sure.
|
|
|
Boffis
International Hazard
Posts: 1852
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Not even Baron von Frankenstein had contemplated such a thing.
Lightening pierced the night, the rumble of thunder echoed off distant peaks and then;
A simultaneous flash of lightening and crack of thunder animated the corona bunds sending frenzied blue streamer racing down the insulator stacks.
A darkness returned that seemed to render impotent the faint glow of the IPad screen. But wait, could it be? Still borne and left for dead for almost
ten years, now it stirred. Yes there was life once again in the old chloranil thread. 

Now for something completely sensible.
Preparing chloranil by treating phenol with HCl and sodium chlorate is not a good way to generate chloranil because the initial product is
2,4,6-trichlorophenol which is then difficult to convert to chloranil, although there is a procedure described by Lübbecke and Boldt (1) for a
similar route using hydrochloric acid and hydrogen peroxide with a large amount of magnesium chloride as a catalyst. I have found that it is better to
start with a para-substituted benzene derivative such as quinol, para-phenylenediamine, p-aminophenol or paracetamol which hydrolyses to the former
compound. That said the original poster was about the nature of the oxidising agent.
I did not try the original posters idea with TCCA or NaDCCA because the large amount of isocyanuric acid formed would be difficult to remove from the
chloranil except possibly by leaching in a soxhlet extractor with acetone. If TCCA or the sodium salt are all that you have available as the
chlorinating agent then it is probably better to generate the chlorine externally. There is a published procedure for the use of this method for the
preparation of o-chloranil from catechol via this route although the final oxidation of tetrachlorocatechol to o-chloranil is done externally (2).
A search of the SM forums reveals many references to the preparation of chloranil, some personal others in journals or patents but very little in the
way of actual personal detailed accounts of its preparation have ever been posted. I hope to remedy this over the coming weeks as I intend to post not
only the preparation but the purification of the product which is usually highly impure. I have made many batches of chloranil over the years and
recently I required rather a lot of it to study some of its products. Historically, I used paracetamol in hydrochloric acid with sodium nitrate as the
oxidizing agent but this is the preparation from hell, monstrous toxic foam belching chlorine, nitrogen oxides, chloropicrin and the rest for a final
yield of only 35-40%. Looking for a cleaner method I decided to try oxidizing quinol (hydroquinone) in hydrochloric acid with various oxidizing agent,
I tried sodium chlorate, sodium chlorite and hydrogen peroxide. The first two worked well though the occasional micro explosions in the vapour above
the reaction mixture when using sodium chlorite are a little unnerving but my attempts with 30% hydrogen peroxide failed.
The failure of the hydrogen peroxide method came as a surprise given the number of patents (3, 4 & 5) that describe various modification to this
method. Has anyone on this forum ever tried the hydrogen peroxide method on any substrate? In the paper by Lübbecke and Boldt (1) that describes this
method they us magnesium chloride as a catalyst. However, in the Lübbecke procedure 270ml of concentrate (presumably 36%) hydrochloric acid are used
for just 1.7g of phenol; admittedly the yield is claimed to be 99%.
I would be interested to hear about other people’s experiences preparing this material. I intend to produce a section in the “Pre-publication
forum” eventually.
(1) Lübbecke, H. and Boldt, P.; Tetrahedron 1978; v34; pp1577 to 1579
(2) US patent 4196132
(3) DE 2645114
(4) EP 0326456
(5) US 5334735
|
|
|
tsathoggua1
Hazard to Others
Posts: 335
Registered: 8-1-2017
Location: Beyond the pale
Member Is Offline
Mood: Phosphorescent
|
|
How about using the reaction between hot, concentrated NaOH or KOH and chlorine gas bubbled through to produce chlorates? been a long time (years)
since that road was travelled personally but seem to remember that if cold, hypochlorites are the result of this, whilst chlorates are the result,
along with presumably some chloride.
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
@Boffis
Did you come to an optimized procedure for producing chloranil from hydroquinone/p-aminophenol? Would be interesting to hear!
[Edited on 21-12-2017 by nitro-genes]
|
|
|
Boffis
International Hazard
Posts: 1852
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
@nitro-genes; I wouldn't say "optimised". I found, as mentioned in the Lübbecke and Boldt paper, you basically need a very large excess of
concentrated hydrochloric acid over the phenolic compound and oxidizer or you get a lot of partial chlorinated products, di and trichlorobenzoquinones
and sometimes chlorinated hydroquinone derivatives mainly. Hydroquinone is definitely "cleaner" in that much less red alcohol soluble "tar" is
produced.
The important thing is the purification because most of the intermediate stages can be recycled into the next batch. I have found that with
paracetamol I get a high apparent yield of deep orange product after the initial cold water washing and drying (the product gives off a very acrid
vapour while drying, possibly occluded chloropicrin). The crude product is then mixed with an equal weight of isopropanol and left to stand for a
short while at room temperature and the then sucked dry on a buchner funnel and washed with a little more isopropanol. This gets rid of most of the
red tarry material and much of the acrid material. The leachate can be "neutralised" with caustic soda solution before disposal.
The product is now usually cadmium yellow and can be further purified with small volumes of hot isopropanol and/or acetone to give a lemon yellow
pearly powder that consist of chloranil and some trichlorobenzoquinone. The later can be leached out with boil water according to a patent or the
whole can be recrystallised from acetone but it requires a lot of solvent. I usually use it as at this stage since most of the products I make are
easily purified. The materials leached from the product in these later stages can be recycled into subsequent batches. Without recycling a 35% yield
of crude chloroanil is pretty reasonable starting with paracetamol.
With hydroquinone I got much less red tar but the overal yield is about the same. Recycling the lower chloroquinones and hydroquinones greatly
improves the overal yield.
Interestingly I eventually purchased some commercial chloranil but have found it to be much less reactive, particularly towards sodium nitrite in the
nitrite substitution route to nitranilic acid.
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Thanks for the info, especially the purification steps are helpful! Been wondering if it would matter if oxidation to benzoquinone or chlorination
would be favourable during the initial stages of the reaction, as most with most available syntheses both seem to occur simultaneously. Can
benzoquinone actually be chlorinated all the way to chloranil? Do the extra chlorine groups attached prevent side reactions, like complete destruction
of the ring e.g? Would it be beneficial to tetra-chlorinate first and form the quinone last?
Attachment: Chlorination of N-acyl Derivatives of p-Aminophenols (Naphthols) and p-Phenylenediamines.pdf (69kB)
This file has been downloaded 445 times
[Edited on 28-12-2017 by nitro-genes]
|
|
|
Boffis
International Hazard
Posts: 1852
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
@nitrogenes, there are plenty of references to the reductive chlorination of benzoquinone with hydrochloric acid to chlorohydroquinone so I have
always assumed that the process simply progresses via repeating this process and oxidation alternately. I have noticed that with insufficient
oxidising agent the product is a colourless crystalline material that I have taken be be various polychlorohydroquinones. A paper was once posted on
this website, I think by Nicodem, from a Korean or Chinese publication (in English) that looked at the isomer make up of some intermediate
chlorination products.
As the chlorination progresses I think it becomes harder to add further chlorines and that this is why the massive excess of hydrochloric acid may be
required, simply to maintain a high concentration of HCl in the latter parts of the synthesis when some of the HCl has been consumed. If this is the
cases it may be better to carry out the reaction in stages. One problem though that I have noticed is the very low solubility of the tri and
tetrachlorobenzoquinones in the aqueous reaction mixture which may result in a "protective layer" effect reducing the efficiency of the
chlorination/oxidation in the later parts of the synthesis. This may be another reason for the very large volume of hydrochloric acid relative to
reactant in the paper by Lübbecke and Boldt. The paper you attached is very interesting indeed since it uses a non-aqueous solvent in which the
products are probably much more soluble.
I am sure you could use benzoquinone and HCl to start with and then revert to an oxidising agent+hydrochloric acid system.
Incidently in the early days I used sodium nitrate and conc. hydrochloric acid, this required a large amount of hydrochloric acid to keep the salt in
solution, it gave better results but the foam from hell and the lethal fumes were a problem.
|
|
|
Boffis
International Hazard
Posts: 1852
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Has anyone on this board ever tried to produce sodium chloranilate or sodium nitranilate? I prepared these compounds long ago but recently repeated
these experiments as part of my ongoing investigation of chloranil chemistry. Both were prepared by reacting chloranil with sodium hydroxide and
sodium nitrite respectively.
When I compared my new sodium chloranilate, prepared from carefully purified chloranil, I noticed that the older crystals were lustrous black and
resemble K permanganate while the new material is a glassy deep maroon red. After some experimentation I discovered that it is best to precipitate
free chloranilic cid and recrystallise it from methanol. The first crop of crystals are brilliant scarlet red needles that contain methanol of
crystallisation that is rapidly lost on drying even at 50 C. When the methanol evaporates the crystals crumble to a terracotta red coloured powder but
I noticed that within this powder are a few blocky deep red lustrous crystals that do not crumble. By evaporating the filtrate down, further crops of
chloranilic acid are obtained but the amount of the mysterious glassy red crystals increases each time. By many careful recrystallisations I have
obtained the two compounds fairly pure. The terracotta coloured dust dissolves in NaOH solution to give deep red sodium chloranilate crystals on
cooling while the red crystals dissolve to give lustrous purplish black crystals on cooling that resemble potassium permanganate. Chromatography
reveals that even the sodium chloranilate prepared from rerystallised chloranil and commercial 98% chloranil gives a fair amount of the second
compound.
Work on the recrystallisation of sodium nitranilate proved even more complex. An examination of the first crop of recrystallised material under the
microscope revealed the presence of 3 substances plus amorphous resin. The sodium nitranilate forms "spear head" shaped rounded tablets or bipyamids,
the second compound forms brighter orange "boat shaped" tablets or short prisms and the final compound a deep red prisms. I have now, through many
recrystallisations separated out pure deep orange sodium nitranilate and the deep red prisms. But the second compound, the "boat shaped" tablets, I
cannot separate from about an equal amount of sodium nitranilate. By precipitating the silver salt of the red prisms, drying it and then decomposing a
known weight suspended in water with hot dilute KI and filtering off the AgI and weighing it I found that the compound has almost exactly the same
silver content as silver nitranilate and is equally explosive (the sodium salt of the red compound is noticeably less enegertic than sodium
nitranilate).
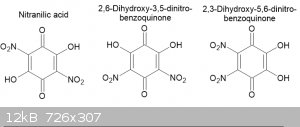
My conclusion is that the red crystals from sodium nitranilate are an isomeric compound and of the 2 possibilities I think that the sodium salt of
2,6-dihydroxy-3,5-dinitrobenzoquinone is more like than the 2,3-dihydroxy-5,6-dinitrobenzoquinone as vicinal dinitro quinones tend to hydrolyse
readily to hydroxy-nitro benzoquinone; hence the reason that nitranilic acid forms in the first place rather than tetranitrobenzoquinone.
This raises the possibility that the red crystals extracted from crude chloranilic acid are also an isomer of it, perhaps
2,6-dihydroxy-3,5-dichlorobenzoquinone.
I can't find any information on the possible isomers of either nitranilic acid or chloranilic acid. What do fellow MS member think? Any suggestion on
how I cound investigate these compounds? And does any one have access to reaxys or similar and would be kind enough to run a search for the isomeric
compounds? The image below shows the three possible nitranilic acid isomers.
|
|
|
Diachrynic
Hazard to Others
Posts: 222
Registered: 23-9-2017
Location: western spiral arm of the galaxy
Member Is Offline
Mood: zenosyne
|
|
I've made sodium nitranilate from chloranil that I prepared by the procedure from Axt, see http://www.sciencemadness.org/talk/viewthread.php?tid=9424#p... and http://www.sciencemadness.org/talk/viewthread.php?tid=9424#p...
The reaction that Axt gives (and was found word for word in a later paper without citing Axt) for sodium nitranilate worked well for me.
I also tried the reaction in acetone as stated in https://www.doi.org/10.5562/cca3333 but that reaction just produced a dark brown tarry precipitate I wasn't able to work up further.
Pictures of the crystals I obtained, left sodium nitranilate and right lead nitranilate:
 
The tetrachloro-p-benzoquinone I made probably isn't completely pure. I did a TLC in petrol ether/ethyl acetate 3/2 on silica 60 F254
plates:
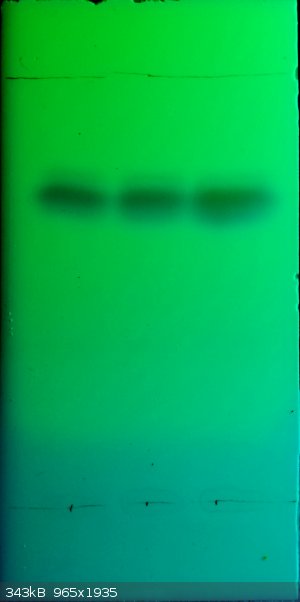
There seen to be at least two other compounds in there...
Purification of the tetrachloro-p-benzoquinone seems like a headache, and seems to involve a lot of steps. See the entry in Beilstein https://archive.org/details/BeilsteinsHandbuchDerOrganischen... and https://archive.org/details/BeilsteinsHandbuchDerOrganischen....
As for chloranilic acid I have no experience with it, but Beilstein gives no isomers apart from 2,5-dichloro-3,6-dihydroxy-1,4-benzoquinone. Beilstein
mentiones that two hydrates of the sodium salt exist, the trihydrate (crystallizes above 35 °C) as black crystals and the tetrahydrate (below 18 °C)
as deep red needles. Maybe that explains the difference in crystals you observed? https://archive.org/details/BeilsteinsHandbuchDerOrganischen...
we apologize for the inconvenience
|
|
|
Boffis
International Hazard
Posts: 1852
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
@ Diachrynic. Thank you for your response. I must admit I don't often think to go to Beilstein but the information on the different hydrates of sodium
chloranilate is very interesting and could certainly account for the different products and possibly also for the free chloranilic acid recrystallised
from methanol. Perhaps the first formed crystals when the solution is still warm do not take on methanol of crystallisation. I am not at home at the
moment but when I am I should be able to test theses ideas.
As for the sodium nitranilate, I prepare it by a method essentially identical to that of Axt but my crystals are not like those you illustrate above,
which look like red prisms. Mine are bright orange bipyramidal crystals or flattened elongate "spearhead" shaped tablets after several
recrystallisation from hot saturated solution. It is possible once again that I am getting different hydrates and that the technique I use where the
hot saturated starts to crystallise while still very hot results in a persistent mixture of hydrates in an almost constant ratio. I will have a look
into this too when time permits. The red needles that I recovered at the end of crystallisation, however, are quite distinct. They dissolve in hot
water to give a red solution as against the yellow orange colour of sodium nitranilate and they are significantly more soluble. I will investigate
this substance again in due course. By the way, sodium nitranilate prepared from crude homemade chloranil is almost always rather brownish sometime
even brownish red due to the presence of a tarry compound. This can't be removed by simple recrystallisation and required charcoal+kieselguhr
treatment while the solution is hot.
The issue of purifying crude chloranil is problematic but I found a couple of interesting paper on the preparation of chloranil, its reactions and
those of trichlorobenzoquinone, the main impurity. I have found from experience that the larger the excess of hydrochloric during the preparation the
greater the proportion of chloranil in the final product. Graebe in his paper reports the presence of about equal amounts of trichloro and
tetrachlorobenzoquinone in the crude "chloranil" but then goes on to describe how to separate them. I have attached a translation of the Graebe paper
as others might be interested too particularly in some of the more unusual derivatives.
Incidentally, the easiest route to pure chloranil free from lower chlorinated compounds and tar is from the old wood preservative sodium
pentachlorophenol, while now banned in Europe it still crops up occasionally on Ebay etc. This compound is easily purified from the crude commercial
product and then oxidised with 40-50% nitric acid. The stronger the acid the higher the yield but even fuming nitric acid only gives 35% were as 40%
acid give 25% yield, the poor yield is compensated for by the simplicity of the procedure and the cheapness of the starting material. The by-products
are things like oxalic acid and an orange tar that is soluble in isopropanol so easily removed. If the free pentachlorophenol is first treated as a
solution in a chlorinated solvent with chlorine so called hexa and heptachlorophenol are formed (actually cyclohexadione derivatives) and these give
almost quantitative yields of chloranil when treated with nitric acid.
Attachment: Investigations into quinone group Aannalen Graebe 1868 English.docx (140kB)
This file has been downloaded 107 times
|
|
|









