Fery
International Hazard

Posts: 1009
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
1,4-naphtoquinone by naphtalene oxidation with dichromate
Here something from literature:
http://www.orgsyn.org/Content/pdfs/procedures/CV4P0698.pdf
https://www.prepchem.com/synthesis-of-1-4-naphthoquinone/
Preparation of 1,4-naphthoquinone
10 g of chromic acid are dissolved in 65 ml of ice-cold 80% acetic acid. A solution of 10 g of naphthalene in 95 ml of glacial acetic acid is then
slowly added, and the whole allowed to stand for three days at the ordinary temperature with occasional shaking. At the end of this time the liquid is
poured into 850 ml of water, and the precipitated 1,4-naphthoquinone filtered off. It is almost pure, but may be purified by sublimation or by
recrystallisation from alcohol. It forms golden yellow plates which melt at 125° C. It possesses a biting odour and, like all true quinones, is very
volatile with steam. Yield about 40%.
Preparation of organic compounds, E. de. Barry Barnett, 137, 1912
https://sci-hub.st/10.1039/CT88139002207
Attachment: japp1881.pdf (381kB)
This file has been downloaded 328 times
This is an experimental work and I'm not telling you to do it this way. I decided to try dichromate instead of CrO3, as I wanted to avoid preparation
of CrO3 from dichromate and I have a lot of very cheap Na dichromate. The final step is not yet done but I post everything which is already done:
------- calculations --------
Na2Cr2O7. 2H2O 298 g/mol
chromic acid 118.01 g/mol
naftalene 128.1705 g/mol
H2SO4 98.079 g/mol
Na2Cr2O7 + H2SO4 -> 2 CrO3 + Na2SO4 + H2O
C10H8 + 2 CrO3 + 6 CH3COOH -> C10H6O2 + 2 Cr(CH3COO)3 + 4 H2O
Na2SO4 + 10 H2O -> Na2SO4.10H2O (below 32,384 C), .7H20 above
Na2SO4 + Cr2(SO4)3 + 24 H2O -> 2 NaCr(SO4)2.12H2O (seems not to occur as the crystals on the flask wall are colorless)
https://doi.org/10.5560/znb.2012-0225
1/2 scale
0,6 mol / 2 = 0,3 mol Na2Cr2O7.2H2O = 89,4 g
0,3 mol H2SO4 = 29,4g 100% = 30,65 g 96% density 1.83 g/ml = 16,75 ml
0,25 mol naftalene = 32,04 g
Na2Cr2O7 solubility in water at 20 C 64,4%
90g salt + 50g H2O
90/140 = 64,3%
----------- experiment --------------
Place 89,4 g Na2Cr2O7.2H2O into RBF reactor and add 50 ml H2O, dissolve as much of dichromate as you can. While swirling in hand add dropwise 31g 96%
H2SO4 (theoretical minimal amount 30,65 g 96% H2SO4 = 16,75 ml), the T raises to approx 50 C (it is possible to touch the flask)
add 75 ml conc. acetic acid
cool to 0 C, while cooling down at 20 C crystallization observed (? Na2SO4.10H2O ?)
dissolve 32,0 g naphtalene in 300 ml of conc acetic acid (requires slightly warming to approx 30-40 C)
added naphtalene in acetic acid dropwise while stirring and cooling with snow-water bath and at such a rate that the T was kept below 15 C (most of
time it was between 5-10 C)
time +0h addition started, T 0 C
time +2h addition finished, T 10 C, snow-salt bath removed and continued stirring, reaction mass quite thick, stirring difficult and at very low RPM
time +3,5h reaction temperature 35 C (mixture much less thick, stirred better than when cold - stirring RPM much higher at first sight), T lowered to
25 C using shalow water-snow bath, then the bath removed and continued stirring
time +4h reaction temperature 35 C
time +4,5h reaction temperature 37 C
time +5h reaction temperature 35 C
time +6h reaction temperature 33 C, flask completely covered with cca 4 layers of old folded t-shirt as a thermal insulation
time +7h reaction temperature 32 C
time +8h reaction temperature 32 C
time +9h reaction temperature 32 C
time +10h reaction temperature 32 C
time +11h reaction temperature 31,5 C
time +12h reaction temperature 31 C
time +18h reaction temperature 27 C
time +24h reaction temperature 27 C, stirring stopped, the T was 7 C higher than lab. temperature very likely due to some heat from stirring motor +
friction when mechanical stirring
time +28h reaction temperature 23 C, thermal insulation removed, reaction mixture mostly green
time +48h reaction temperature 20 C
reaction allow to stand for 5 more days occasionally stirring for 1 minute few times per day (cca 5x) using magnetic stirrer
temperature inside lab 20 C
then pour into 3 L of cold stirred water
vacuum filter using sintered glass funnel (sinter No. 3 used, vacuum at least to half of atmospheric pressure, the filtration lasted very long time
for a lot of hours)
the obtained yellow paste mas was mixed well with 100 ml of cold water in a beaker using thick glass rod and filtered again using the same funnel, at
the end the filtrate was almost colorless
the product in a form of a paste dried over anhydrous CaCl2 for more than 1 month, it did dry to some extent but did not dry out completely
divided into 5 g portions (totally 35 grams)
each 5 g portion extracted 3 times with 50 ml medicinal petrolether (fraction boiling 40-60 C in which naphtalene contaminant does not yet melt as its
m.p. 80 C when pure), the mother liquor was reused for forever, just always adding small amount which was lost... 2 Erlenmayer flasks used, 100 ml for
extraction and 50 ml for crystallization, both closed with piece of plastic foil and rubber band to reduce solvent loses, crystallized at -18 C in
fridge for 1 hour, total consumption of solvent 100 ml due to evaporation loses, solvent/mother liquors always only decanted between the 2 flasks,
never filtered (decantation was OK, filtering really unnecessary)
that way 1,4 napthoquinone contamined with unreacted naphtalene extracted and separated from red/brown sideproducts
++++++++++ this is not worth of repeating +++++++++++++
but I post it here as someone could have the same idea as me that something could be obtained from the voluminous green solution by steam distillation
the sintered glass funnel washed with 2 x 10 ml of ethanol and the washings added to more than 3 L of filtrate to destroy possible remainders of
chromates and then steam distilled in a lazy way - using simplee distillation apparatus WITHOUT extra source of steam
the more than 3 L of filtrate was distilled and 800 ml of distillate collected, it had yellow color, a little of maybe naphtalene separated out of it
which was filtered out
the yellow 800 ml solution was extracted 3 x 50 ml of ethyl acetate and the organic layer was let to evaporate outside - the ester extracted some
acetic acid too so after it evaporated there was dark solution of some unknown compound in acetic acid, the acid later slowly evaporated leaving a
little of dark brown crystals (cca 100 mg)
info concerning solubility:
https://pubchem.ncbi.nlm.nih.gov/compound/1_4-Naphthoquinone...
In water, 0.35 g/100 ml at 25 °C
INCHEM-IPCS; International Programme on Chemical Safety (IPCS), ICSC:1547. Available from, as of Jan 1, 2006: http://www.inchem.org/
info how to separate unreacted naphtalene from 1,4-naphtochinone using 50% acetic acid, this seems to be the same document as the above attached
japp1881.pdf file:
https://pubs.rsc.org/en/content/articlelanding/1881/CT/CT881...
pictures follow:
dichromate in RBF

dichromate almost dissolved in minimal amount of H2O

dropwise added H2SO4 while swirling in hand, the reaction Na2Cr2O7 + H2SO4 -> 2 CrO3 + Na2SO4 + H2O, the T raised to approx 50 C so it was still
possible to touch the flask by hand

addition of solution of naphtalene in acetic acid while stirring and cooling in snow-water bath
      
here it is visible that the small T difference between inside of flask and room temperature which caused vapor to condense on the walls of flask and
wash out the orange-green dichromate / CrO3 / chromium acetate from the colorless sodium sulfate so at the end only colorless Na2SO4.10 H2O crystals
stayed on the wall of flask
  
thermal insulation of the reactor using an old t-shirt which was degraded to be used for at most wiping the floor prior definitely be trashed and here
it still greatly served for a high science instead
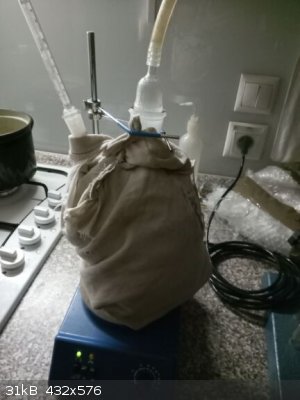
  
vacuum filtering, very tedious and long lasting process of the paste-like product
 
drying over CaCl2, it dried only partially after 1 month

extraction with petrolether, on the left in the beaker the remainder, in the 2 Erlenmayer flasks the product extracted (1,4-naphtoquinone contamined
with unreacted naphtalene), it has to be purified but I do not know when I have a time to do it (perhaps recrystallization from petrolether or
extraction of naphtoquinone with hot 50% acetic acid)

[Edited on 12-1-2021 by Fery]
|
|
|
Fery
International Hazard
Posts: 1009
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
now the part which is not worth of repeating, seems some polynaphtols obtained in tiny amount, maybe something like juglone from husks of walnuts
(Juglans nigra, Juglans regia)
possible dichromate / CrO3 in filtrate destroyed by ethanol and then performed lazy steam distillation (just simple distillation without extra source
of steam)
  
a little of something like naphtalene or naphtoquinoe floating on the surface of distillate which was removed and which sublimed away in few days
  
the 800 ml distillate extracted 3 times with 50 ml of ethyl acetate, this is the first extraction

the 3 extractions collected together, let to evaporate outside of building for few days

the ethyl acetate extracted not only the colored compound but also some of acetic acid so after the ester evaporated, the dark red-brown colored
compound stayed dissolved in acetic acid and the acid evaporated only very slowly at room temperature during few days

very tiny amount of an uknown compound obtained, cca 100 mg - so not worth of repeating this part of the experiment, it just proved that there is not
too much to be steam distilled from the voluminous green filtrate
  
|
|
|
Boffis
International Hazard
Posts: 1852
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Interesting work Fery, and a nice write-up even if not 100% successful! I had been thinking about trying this reaction but you seem to be struggling
so I might wait and see how thing work out with yours!  . .
I wonder if there is too much water in the original mixture from the reaction of sodium dichromate and sulphuric acid. I read somewhere that CrO3 is
made from dichromate and excess warm sulphuric acid. The CrO3 then separates as a heavy immiscible liquid (immiscible with the excess sulphuric acid
alkali hydrogen sulphate mix) and can then be separated and cooled. I wonder if you can do this and then mix the liquid CrO3 with the acetic acid
directly giving you better control of the water.
Your filter cake looks a bit pale I was under the impression that 1,4-naphthaquinone was bright yellow. I wonder if you could recrystallise it from a
more water tolerant solvent such as ethanol, isopropanol or acetone first to separate out some of the naphthalene without having to dry the volatile
quinone first. Or alternatively dissolve in a solvent in which it has good solubility at room temperature and then dry the solution with MgSO4 or
Na2SO4.
|
|
|
Fery
International Hazard
Posts: 1009
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
Hi Boffis, the most important part is still waiting - purifying the yellow-orange crystalline mass in the 2 small Erlenmeyer flasks, it should be a
mixture of the 1,4-napthoquinone with some unreacted naphtalene. You inspired me to try this synthesis when you mentioned benzoquinone for Diels-Alder
reaction with sorbic acid here:
https://www.sciencemadness.org/whisper/viewthread.php?tid=15...
After reading your post I had an idea for what to finally use my naphtalene and dichromate.
The original procedure which uses CrO3 uses cca 80% acetic acid + 20 % H2O to dissolve CrO3 and then 4-fold amount of acetic acid is added in which is
the naphtalene dissolved. In my reaction mixture there was somewhat more of H2O but not too much. If the original procedure uses totally 400 g of
acetic acid and 15 g of water and mine 400 g of acetic acid and 60 g of water it should not be a big difference for the oxidation, naphtalene could be
somewhat less soluble but as oxidation proceeds and naphtalene is consumed more of it dissolves in the acetic acid. Probably the lower solubility of
naphtalene in my acid made the oxidation slower and easier to control - maybe it crystallized out first as unreacted and later as it was consumed in
the reaction the previously crystallized naphtalene redissolved... At the end the reaction mixture is poured into huge excess of water anyway to
precipitate product. I was more concerned about Na2SO4, I expected it to crystallize and make stirring impossible but that did not happen, albeit the
stirring was somewhat slow at the beginning but magnetic stirrer was able to rotate and later the mixture was even better stirrable.
Yes the filter cake was quite pale, very likely due to very small particles - strange how dark residuum was obtained from it - the dark brown mass in
the small beaker on the left side of the 2 Erlenmayer flasks with the product. Maybe there could be isolated the same compound from it as I obtained
by steam distillation - just guessing according its color...
[Edited on 13-1-2021 by Fery]
|
|
|
Boffis
International Hazard
Posts: 1852
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Hi Fery, did you get any further with the purification of your naphthoquinone? I would also be interested to see how you got on with a Diels Alder
condensation. I tried several experiments with different ratios of benzoquinone to sorbic acid as benzoquinone has at least a theoretical possibility
of a double condensation. Most of the experiments yielded a small amounts of beautiful pale yellow diamond shaped tablets and a black crystalline
quinhydrone type material. The best yield of the yellow compound which I take to be 5-carboxy-8-methyl-octohydro-napthoquinone was from the "slow
bake" process. The solution of sorbic acid and benzoquinone in benzene was sealed in a 100ml schott bottle and place on the back of the central
heating boiler (about 40-45 C) for about 4 days. Since a lot of unreacted starting compounds were recovered I think this reaction would require about
1 month under these conditions. I may try it again and also with refluxing toluene.
By the way I found that all of my sorbic acid batches (including commercial material and material I extracted from K sorbate is contaminated with a
sparingly soluble material and so needs either soxhlet extraction with benzene or recrystallisation from methanol (about 8ml per gram, treat with
keiselguhr or similar filter aid and then evaporate the filtrate down to half volume; you can use 4 ml but then you can't filter it fast enough to
stop it crystallising in the funnel).
[Edited on 15-2-2021 by Boffis]
|
|
|
Fery
International Hazard
Posts: 1009
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
Hi Boffis, it is still a mixture of unreacted naphtalene with 1,4 naphtoquinone, nice yellow color. I still do not know whether to use 1:1 acetic acid
: H2O method or just recryst from hexane (I expect naphtalene better soluble in hexane than 1,4 naphtoquinone).
Maybe I can let it react unpurified (naphtalene wouldn't react)... I have maybe 2 kg of sorbic acid and maybe 1,5 kg of K sorbate. I've also bought
maleic anhydride for Diels-Alder. But did not yet try anything.
|
|
|
Boffis
International Hazard
Posts: 1852
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Hi Fery, have you tried using methanol or ethanol diluted with a little water, I have found that some quinones are pretty soluble in simple alcohols
while naphthalene is less so.
During my search for papers on Diels Alder reaction I came across a few papers about Lewis acid catalyzed Diels Alder reactions and I was wondering if
AlCl3 might assist this reaction when carried out in benzene. The reaction seems very slow even at 80C and I can only find evidence if a 1:1 addition
product.
|
|
|
Fery
International Hazard
Posts: 1009
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
Hi Boffis, I have plenty of anhydrous AlCl3 (thx to chemship1987). I just have to purify the product, it seems to contain naphtalene (or could be the
scents of naphtalene and 1,4-naphtoquinone very similar?). I prefer solvent where naphtalene well soluble and naphtoquinone less soluble so I think I
will use medicinal petrolether (isohexanes fraction). I would certainly prefer to use AlCl3 to speedup Diels-Alder reaction, thx for the hint.
|
|
|
Boffis
International Hazard
Posts: 1852
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Hi Fery, I'll try and find some of those papers on Lewis acid assisted Diels Alder reaction.
I am just about to kick off a 0.1 molar scale 1:1 condensation in refluxing toluene.
|
|
|
|









