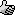| Pages:
1
2
3
4 |
Jor
National Hazard

Posts: 950
Registered: 21-11-2007
Member Is Offline
Mood: No Mood
|
|
Very, very nice. You should try the barium-salt as well. not_important gave a good idea on how to prepare it.
|
|
|
woelen
Super Administrator
Posts: 8011
Registered: 20-8-2005
Location: Netherlands
Member Is Offline
Mood: interested
|
|
I still think that I have K3CrO8, but the crystals are larger. If the material is crushed, then it becomes brown. This also is what YT2095 has noticed
already. I have another sample, with smaller crystals and that indeed is somewhat more brown:
The compound, from a previous batch:

The last batch:

I also noticed the difference between my two samples, but on further testing I can only conclude that they are the same compound. Both give a
red/brown solution, both give a blue solution on acidification, and both violently explode on heating, giving exactly the same kind of yellow smoke.
When mixed with S it behaves like flash, with red P it detonates.
So, I am quite confident that they are the same compound, but either in a different crystal shape, or a different purity. Actually, I have the
impression that the black material is more pure. These crystals really are nice and glittering, while the brown material has more dust-like stuff
around the crystals. Actually, the crystals are not really black, I already wrote in my first post in this thread that they are very dark brown.
A similar thing I know from anhydrous copper(II) chloride. This stuff always is described as yellow/brown. I once made this, and I obtained dark
chocolate brown crystals. On adding them to water they nicely gave a clear green/blue solution and on crushing them, I obtained a yellow/brown powder.
So, again I think that the situation here is similar. Large crystals look darker.
The experiment with ammonium peroxochromate looks very interesting. I knew of the ammine complex and never dared making more than a few mg of that,
but the ammonium salt of the peroxo chromate ion sound very good. I'll definitely try that one myself.
I do not regret that I made this compound. It actually adds a nice chemical to my set of chemicals. And yes, you have to be careful. I also mentioned
it on my web page, you really have to be careful for the spraying droplets of solution. Chromates, and also perchromates are toxic carcinogens. But
there are nastier chems.
EDIT: There is one difference between both of my samples and the sample of Taoiseach. My samples explode on heating, even when not confined and not
mixed with any other chemical. I never could get it to burn, it explodes, as shown on my website.
YT2095, could you put a tiny(!!) amount in a test tube and heat that above a flame? Does it burn, or does it explode with a POP sound? Don't do this
test in open air, use a wide test tube. It still is unconfined in that case, but no K2CrO4-smoke is spread in the air.
This is another page on K3CrO8 I have written some time ago.
http://woelen.scheikunde.net/science/chem/exps/raw_material/...
It also contains a synth method, much like the one, presented here. I have done that synth two times, but one time I had a runaway, even from ice cold
material. Hence my new method which uses much more dilute H2O2 (it is based a method from Edel, who uses even more dilute H2O2).
As you see, interesting material! I certainly will go more into it and the contrinution of Taoiseach is appreciated very much  . Good to see that this stuff raises so much interest! . Good to see that this stuff raises so much interest!
[Edited on 17-3-08 by woelen]
[Edited on 17-3-08 by woelen]
|
|
|
UnintentionalChaos
International Hazard
Posts: 1454
Registered: 9-12-2006
Location: Mars
Member Is Offline
Mood: Nucleophilic
|
|
Potassium tetraperoxochromate (V)
Woelen, this is an excellent synthesis of a very interesting compound. I'm at college or I'd perform the experiment. Problems I do see are with your
page explaining it though, mostly problems with redox. As you said yourself, it would probably be wiser to write the formula as K3Cr(O2)4
Now I call attention to your reaction formula:
2CrO4(2–) + 7H2O2 + 2OH(1)– → 2CrO8(3–) + 8H2O
You are making 2 equivalents of an anion containing 4 peroxo ligands. Those 8 peroxo groups have to come from somewhere, but you've only added 7
equivalents of H2O2 (The source of peroxo ligands)
The other thing that worries me is the oxidation state of the chromium. In chromate/dichromate, you have hexavalent chromium. In Potassium
tetraperoxochromate, the chromium is pentavalent. The only available reductant is H2O2. A quick google search yields quite a few hits on the reduction
of Cr(VI) by H2O2, ultimately leading to Cr(III). The low temperatures in the synthesis likely slow the reaction down so the product can crystallize
out before being reduced or rather, that further reduction is extremely slow or negligible at the reaction temps. The half reactions are as follows:
Cr (VI) ---> Cr(V) - e-
2OH(1-) + H2O2 ---> O2 + 2H2O +2e-
Either 2 Cr(VI) ions are reduced by each molecule of hydrogen peroxide or this half reaction might (probably) occur:
OH(1-) + H2O2 ---> HO2 +H2O + e-
The hydroperoxide radical could then go on to react with either more Cr(VI) and be oxidized to oxygen gas or probably with more hydrogen peroxide...
DISCLAIMER: I have no formal or even informal knowledge of radical chemistry so forgive any gross errors. I'm just going with what I know about redox
chemistry. I have requested some reading material on the reactions of Cr(VI) with H2O2 and will report back once I've read them.
There will always be some bubbling as some H2O2 must eventually be oxidized to oxygen gas in order to reduce the Cr(VI). Your reaction equation also
lacks O2 in the products.
Assuming what I just said is correct, you then need to use up another 4 equivalents of H2O2 to get from CrO4(3-) to Cr(O2)4(3-) The lost oxide ligands
match up with the hydrogen lost from H2O2 so you get 4 H2O out of the deal.
So overall, if you assume each H2O2 converts 2 Cr(VI)
2CrO4(2-) + 9H2O2 + 2OH(1-) ---> 2Cr(O2)4(3-) + 10H2O +O2
That would be the simplest possible reaction equation. Now, I move on to combustion.
I requested a paper in Wanted references and needed translations (3):
Burning in potassium tetraperoxochromate mixtures
A. I. Lesnikovich, S. V. Levchik and K. K. Kovalenko
Journal Combustion, Explosion, and Shock Waves Volume 24, Number 4, Pages 458-460, 1988
It can be downloaded from page 18 if anyone is interested.
After the image of the test tube with post explosion products in it, the page reads "After the explosion, the test tube is filled with a yellow smoke,
which mostly is K2CrO4, and K2O." According to the above paper and my own redox calculations, it should be potassium superoxide (KO2), not potassium
oxide (K2O). Half reactions are as follows.
Cr(V) ---> Cr(VI) + e-
O2(2-) ---> 2O(2-) - 2e-
O2(2-) ---> O2(1-) + e-
O2(2-) ---> O2 + 2e-
If the products contain CrO4(2-) the first half reaction has occured once and the second twice. We are left needing 3 more electrons on the right to
balance, and have used up 2 of 4 peroxo ligands per chromium and 2 of 3 potassium cations have gone to the chromate. Thus, the third and fourth half
reactions must occur once each, giving the 3 needed electrons and keeping the charge neutral by using up the remaining potassium cation. Granted, it
is possible that the KO2 has decomposed in whole or part due to the heat generated by the explosion. Have you tried adding water to the products,
converting any superoxide to hydrogen peroxide which then will reduce Cr(VI) to Cr(III)?
Taoiseach may have quite a point when he voices how dangerous these compounds are.
http://www.pubmedcentral.nih.gov/picrender.fcgi?artid=156738...
This research as well as quite a few papers that I don't have access to seem to suggest that the reason hexavalent chromium is carcinogenic is that is
is reduced to pentavalent chromium in the body, which is what does the real damage. You are possibly holding a jar of pre-prepped carcinogen which
would make inhalation an extreme hazard. Of course the peroxo ligands would be freed in an acid environment, converting it to Cr(III) rapidly. I am
unsure if I would call it more dangerous than hexavalent chromium, but I'd advise treating it with a great deal of respect like all other chemicals.
One last observation. The frozen reaction mix of Taoiseach probably results in extremely small crystals due to intrusion of the ice crystals, whereas
Woelen's liquid reaction mix allows large crystals to grow and appear a different color, sort of like how copper acetate hydrate looks almost black as
bulk crystals but is bright blue green when ground. Is it possible that Woelen's crystals explode when heated simply due to their size? The
decomposition products include a gas...could the crystal size be essentially self-confining?
Anyway, excellent topic Woelen, excellent prep, and excellent site.
[Edited on 3-17-08 by UnintentionalChaos]
Department of Redundancy Department - Now with paperwork!
'In organic synthesis, we call decomposition products "crap", however this is not a IUPAC approved nomenclature.' -Nicodem
|
|
|
woelen
Super Administrator
Posts: 8011
Registered: 20-8-2005
Location: Netherlands
Member Is Offline
Mood: interested
|
|
UnintentionalChaos, very good and deep correction!
UnintentionalChaos, you are absolutely right 
I have regarded the bubbling of oxygen throughout the reaction simply as a side reaction of decomposition of hydrogen peroxide. During the entire
reaction, bubbling continued and I simply was thinking it was decomposition of hydrogen peroxide, nothing more. Stupid me  . .
I already was wondering why this happended, even at -15 C in the refirigerator.
I certainly will change this in my webpage. The nasty thing about peroxide reactions is that stoichiometry of these reactions cannot simply uniquely
be determined by balancing elements and charges, left and right. A similar problem occurs when one is writing down the equations for reduction of
permanganate by hydrogen peroxide.
This indeed is the correct equation: 2CrO4(2-) + 9H2O2 + 2OH(-) ---> 2[Cr(O2)4](3-) + 10H2O +O2
The nasty thing of this equation is that it is the addition of the following:
2CrO4(2-) + 7H2O2 + 2OH(-) ---> 2[Cr(O2)4](3-) + 8H2O
2H2O2 --> 2H2O + O2
The latter is simple decomposition of hydrogen peroxide. And that is where I made a mistake.
The problem can be resolved by assuming preservation of ligands for part of the H2O2:
H2L, L = [OO], L is preserved and we write CrL4(3-) for the peroxochromate ion and H2L for the part of the hydrogen peroxide which goes into ligand
transfer.
The other assumption needed is pure reduction and no decomposition of H2O2. Another preservation of groups needs to be introduced:
H2Ox, Ox = [O2], Ox is preserved in reduction of hydrogen peroxide: H2Ox --> 2H(+) + Ox + 2e. We write H2Ox for the part of hydrogen peroxide,
which is reduced.
Now we have to solve the equation
CrO4(2-) + H2L + H2Ox + OH(-) --> CrL4(3-) + H2O + Ox
Now, using preservation of L and Ox, the equation can be uniquely determined and this is exactly your equation:
2CrO4(2-) + 8H2L + H2Ox + 2OH(-) --> CrL4(3-) + Ox + 10H2O
So, the trick is group preservation of L and Ox.
UnintentionalChaos, thanks for your deep correction. Without this it would have remained erroneous forever! Tonight I'll change this and I'll also
change some other webpages, which deal with the K3CrO8 compound.
[Edited on 17-3-08 by woelen]
|
|
|
woelen
Super Administrator
Posts: 8011
Registered: 20-8-2005
Location: Netherlands
Member Is Offline
Mood: interested
|
|
The other remark, made by UnintentionalChaos about the reaction products of the explosion of K3CrO8 also most likely is correct. I write on this
webpage http://woelen.homescience.net/science/chem/exps/raw_material... , that bubbles of oxygen are produced, when the smoke is mixed with water. This
can nicely be explained when one assumes production of superoxide (or peroxide) of potassium in the explosion. Only a small amount of oxygen is
produced, but it was clearly visible. I'll also change that bit of text.
Edit(woelen): Made link work again.
[Edited on 30-7-16 by woelen]
|
|
|
YT2095
International Hazard
Posts: 1091
Registered: 31-5-2003
Location: Just left of Europe and down a bit.
Member Is Offline
Mood: within Nominal Parameters
|
|
| Quote: | Originally posted by woelen
YT2095, could you put a tiny(!!) amount in a test tube and heat that above a flame? Does it burn, or does it explode with a POP sound? Don't do this
test in open air, use a wide test tube. It
|
I get a sharp Pop and the same yellow "gas" condensate, it will even do that in the open when a small amount is put onto a hotplate, and if Sprinkles
you get a series or tiny crackle like pops.
I must admit though, when I was making mine I didn`t get much in the way of bubbles or foam or Anything in the beaker that was in the fridge, there
was some small activity the next morning when I took it out, I found the reaction to be quite well behaved.
\"In a world full of wonders mankind has managed to invent boredom\" - Death
Twinkies don\'t have a shelf life. They have a half-life! -Caine (a friend of mine)
|
|
|
woelen
Super Administrator
Posts: 8011
Registered: 20-8-2005
Location: Netherlands
Member Is Offline
Mood: interested
|
|
@YT2095: Thanks for your trial. Good to read that you also have the sharp pop and explosion. As UnintentionalChaos mentioned, it must have to do with
crystal size. I'll try crushing some of the crystals and heating that in a test tube. The larger size of the particles most likely both explains the
darker color of our product, compared to Taoiseach' product, and the fact that he gets a fire, while ours explodes.
| Quote: | | I must admit though, when I was making mine I didn`t get much in the way of bubbles or foam or Anything in the beaker that was in the fridge, there
was some small activity the next morning when I took it out, I found the reaction to be quite well behaved. |
The reaction also was quite well behaved for me, but it sure did bubble. I had many tiny bubbles during the entire period that the
reaction was running. So, for anybody doing the reaction, PLEASE COVER UP THE BEAKER by filter paper, a piece of glass or use a loosely capped bottle.
I used filter paper, and the paper was brown at the entire circle, where it covered the beaker.
Twenty years ago, I did the same reaction, but at that time I simply dumped 40ml of 30% H2O2 in the mix. A dark red/brown fountain of hot foam was
spewn out of the beaker, almost 1 meter high  and there was a lot of steam.
Wooo that was frightening. Hence my modification to use dilute H2O2. and there was a lot of steam.
Wooo that was frightening. Hence my modification to use dilute H2O2.
|
|
|
YT2095
International Hazard
Posts: 1091
Registered: 31-5-2003
Location: Just left of Europe and down a bit.
Member Is Offline
Mood: within Nominal Parameters
|
|
the bit I don`t understand is that he said it`s Impact sensitive, and yet despite Several attempts on my part it failed to do Anything at all?
my tests consisted of ~1mg of crystals in very thin paper, then placed on a Very heavy MOT, then a 6 inch nail placed over the paper packet and then
the pointy end of the nail struck repeatedly with a hammer.
I even tested the now VERY flat packet after and it burned up quickly with a purpleish flame, just to make 100% sure it Didn`t "Go off" silently.
also, Try that GAA experiment, it`s quite strange, a great Purple color and very little in way of Fizzing, but Rapidly decomposes when you add water
to it.
\"In a world full of wonders mankind has managed to invent boredom\" - Death
Twinkies don\'t have a shelf life. They have a half-life! -Caine (a friend of mine)
|
|
|
woelen
Super Administrator
Posts: 8011
Registered: 20-8-2005
Location: Netherlands
Member Is Offline
Mood: interested
|
|
The impact test requires Al-foil instead of paper. Paper is fairly soft and dampens the impact strength quite a lot. I'll use a metal block on a
concrete tile. I put 50 mg of material in Al-foil on the metal block and then hit it with a heavy flat metal hammer.
Tonight I'll try the GAA experiment. I sounds like some complex is formed. On addition of water, chromium(III) is formed and this certainly forms a
complex with acetate. Grey color actually is quite common for chromium(III) and I have encountered that before in other reactions, e.g. reduction of a
solution of K2CrO7 in dilute HCl with ethanol or methanol. Also try reduction of potassium dichromate with a warm solution of oxalic acid. That also
gives a deep purple color. Chromium is known for formation of many complexes, especially the metal in the +3 oxidation state, and to a lesser extent
the metal in the +6 oxidation state.
|
|
|
YT2095
International Hazard
Posts: 1091
Registered: 31-5-2003
Location: Just left of Europe and down a bit.
Member Is Offline
Mood: within Nominal Parameters
|
|
the reason I didn`t use Alu is that a "thermit" type reaction could take place doing that.
and that could give a false reading of sensitivity.
the paper I used was the VERY thin cigarette rolling papers (Rizla Silver pack).
edit: I think the bad burn you had on your finger some time back is testamony to it`s reactivity with alu 
[Edited on 17-3-2008 by YT2095]
\"In a world full of wonders mankind has managed to invent boredom\" - Death
Twinkies don\'t have a shelf life. They have a half-life! -Caine (a friend of mine)
|
|
|
woelen
Super Administrator
Posts: 8011
Registered: 20-8-2005
Location: Netherlands
Member Is Offline
Mood: interested
|
|
Again a few hours in the lab
I have tried to repeat some of the results of Taoiseach. First, I made some Cr(NH3)3(O2)2 (it is not Cr(NH3)3(O2)4, but Cr(NH3)3(O2)2). This worked
well at the test tube scale. A webpage on this will follow in due time, but in short, I did the following:
- take a spatula full of ammonium chromate (250 mg or so)
- dissolve in as little as possible of ammonia (20 .. 25%)
- chill to a temperature between 5 and 10 C.
- take 3 ml of 30% H2O2 and chill between 5 and 10 C
- add the hydrogen peroxide in small amounts, assuring that the liquid does not warm up, by swirling the test tube in cold water.
- when all hydrogen peroxide is added, let stand in cold water of appr. 10 C.
After half an hour, quite some red/brown solid was obtained. This is washed once with some distilled water, twice with ethanol and twice with diethyl
ether.
No effort was made to get a good yield, only goal was around 100 mg of material and that succeeded. Final yield was approximately 150 mg of a very
fine crystalline powder. Pictures and webpage will follow.
This material is close to what Taoiseach describes. It is very easily ignited and burns very fast, almost like flash powder. No reductor or other
chemicals added, just the dry compound.
--------------------------------------------------------------------------------
I also tried finely crushing the K3CrO8 and igniting that. I did not succeed in getting it to burn. Moderately crushed powder, fine powder, very fine
powder, it always explodes when heated, even when in the open.
Finally, I tried mixing with 25% K2CrO4 and making a fine powder of this mix. And yes, that burns! It burns fast, giving a purplish/white flame. The
mix with K2CrO4 still is very energetic, exploding with red P and flashing with sulphur.
I have the impression that the material of Taoiseach is a mix of K3CrO8 and K2CrO4. This also may attribute to the lighter color he reports. But, when
the black crystals are crushed, it also becomes much lighter.
--------------------------------------------------------------------------------
I also tried the (NH4)3CrO8. I have severe doubts after several tests. My material does not differ that much from my Cr(NH3)3(O2)2. I noticed that
ammonium chromate is not really soluble very well in ammonia 25%. My spatula full of solid (appr. 250 mg) did not even dissolve in 4 ml of ammonia. I
have the strong impression that at lower temperature no (NH4)3CrO8 is formed, but that a mix of ammonium chromate and the triammine diperoxo complex
is separated from the liquid. My material is quite tame, and this corresponds to the properties of ammonium chromate. Ammonium chromate is more tame
than ammonium dichromate, but a mix of ammonium chromate and the triammine diperoxo complex of course is more energetic again.
So, I think that the lower the temperature, the more ammonium chromate in the mix and the less energetic the compound. This also perfectly well
explains the smell of ammonia when NaOH or KOH is added.
-------------------------
Summarizing:
1) The triammine peroxo complex can be reproduced.
2) The lighter K3CrO8 could well be a mix of pure K3CrO8 and K2CrO4
3) I don't believe that (NH4)3CrO8 is made. I think I and Taoiseach obtained a mix of Cr(NH3)3(O2)2 and (NH4)2CrO4.
-----------------------------------
I did not yet do a hammer test. The weather is rainy and windy, not the type of weather, suitable for working with energetic materials outside.
-------------------------------------
@UnintentionalChaos: The webpage is modified, and your comments are used for improving the page.
[Edited on 17-3-08 by woelen]
|
|
|
len1
National Hazard
Posts: 595
Registered: 1-3-2007
Member Is Offline
Mood: NZ 1 (goal) - Italy 1 (dive)
|
|
I was reading yesterday several studies. A study of 322 workers at a chromium plating plant reveal a 24% death rate from cancer, with 65% being lung
cancer. This is a huge statistical variation from the mean, which I think proves beyond doubt that Cr6+ is carcinogenic. This is not a study, such
as done to prove the carcinogenicity of benzene, at huge doses and with cancer prone mice. This is much more real.
So I wonder what effects burning Cr6+ compounds are likely to have on your health. Even working with dichromates you are likely to inhale some
particles of dust.
I did this reading preliminary to doing some chrome plating, after I noticed that even opening a container of CrO3 you could detect a definite acidic
smell. I have put the plating idea aside now.
I guess the school days demonstration of an ammonium dichromate volcano was really an exercise in cancer innoculation. And I remember many
dichrromate oxidations we did at high school as well. I wonder if anyone can sue the school?
[Edited on 19-3-2008 by len1]
|
|
|
chemoleo
Biochemicus Energeticus
Posts: 3005
Registered: 23-7-2003
Location: England Germany
Member Is Offline
Mood: crystalline
|
|
Come on these are workers do their job at least 8 hours every day, being exposed to Cr6+ aerosols, dusts and so on, for probably dozens of years!
This is no way comparable to a single or quadruple exposure to Cr6+ from school experiments, nor the occasional exposure of Cr6+ in home experiments.
Did you look up, by the way, the normal death rate of cancer in un-contaminated individuals?
Please check this http://info.cancerresearchuk.org/cancerstats/mortality/age/?...
| Quote: | In 2005 cancer was responsible for more than one in three (37%) deaths in people aged under 65 years in the UK. In females under the age of 65 cancer
causes 47% of deaths, while in males it is only 31%.
In people under the age of 75 years, deaths from cancer outnumber deaths from diseases of the circulatory system a, including heart disease and
stroke, and the respiratory systemb combined. |
So in the UK, people exposed to the normal environment die at 37% (1/3rd) from cancer, whilst only 24% of [Cr6+] -exposed die from cancer?
Does this mean Cr6+ is an anti-cancer drug, saving an estimated 13 % of potential cancer patients? 
Let's not get too excited about Cr6+!
Also let's not forget the benzene we happily fuel every few days, straight into our cars! Noone bats an eyelid there!
<back on topic>
Never Stop to Begin, and Never Begin to Stop...
Tolerance is good. But not with the intolerant! (Wilhelm Busch)
|
|
|
len1
National Hazard
Posts: 595
Registered: 1-3-2007
Member Is Offline
Mood: NZ 1 (goal) - Italy 1 (dive)
|
|
I realised I should have clarified after I posted - its not the 24% figure thats outstanding - its the 65% incidence of lung cancer thats well beyond
statistical doubt.
In comparing the 24% cancer rate with the 37% overall death rate from cancer for age<75 we are not comparing like with like, because only
something like 50% of the sample had died.
About benzene, I agree, I indeed stated the data for chromium is a lot more compulsive. Benzene is about 2% of petrol - it would cost a lot to remove
this residual due to the chemistry of cracking, benzene is just a very stable molecule so it has a propensity to be formed. Be that as it may, its in
the lighter boiling fraction - so you get a good doze of it at the bauzer. Yet its sale in pure form is highly restricted due to our legislators
'care' for our health. So its OK to care while it costs them nothing, but the 'care' stops where the money begins. Typical human nature.
See the Carcinogenic effects section
Read section 4.2.1
The reassurance that all these chromium plater deaths are due to people working years in obnoxious conditions is good, however we dont know this for
certain. Its possible they didnt even know they were breathing CrO3 dust. If you look at the CrO3 data measured at the works its of the order of
50ug/m3 which is very small.
[Edited on 19-3-2008 by len1]
|
|
|
woelen
Super Administrator
Posts: 8011
Registered: 20-8-2005
Location: Netherlands
Member Is Offline
Mood: interested
|
|
I have to agree with chemoleo. Even if the CrO3 concentration in such a work place is low, if you are working with the stuff in large quantities every
day, year after year, then you WILL get contaminated, no matter how much you do your best to stay clean.
In my experiments with the chromate and peroxochromate, I tried to be as careful as possible (such as using tissue paper over beakers with the
bubbling solution, and only burning/exploding small quantities of the stuff inside test tubes). Of course, I might have inhaled a few ug of dust-like
hexavalent chromium, but even if this has a similar impact on health as smoking a few cigarettes (which I doubt, given the very small amounts), I
still do not worry. This is only a single set of experiments with a few hours of labwork with hexavalent chromium and now it might be that I don't
touch the dichromates for weeks anymore. I may be exposed to them for a few hours per year, the workers in the plating factories have a similar
exposure every day!
|
|
|
len1
National Hazard
Posts: 595
Registered: 1-3-2007
Member Is Offline
Mood: NZ 1 (goal) - Italy 1 (dive)
|
|
Thanks for the reassurance Woelen. I guess I can rest easy about smelling that CrO3 for a second, if youre prepared to work with the stuff for a few
days. Still chemistry for me is something to make life worth while not to end it early. So its no chromium plating, and if I do any oxidations it
will be in a fume hood with a gas mask (for you still have to open the hood when pouring the stuff). I suggest people burning Cr6+ wear gas masks.
The reasoning is simple - all cancers are the posterity of one initial cancer cell. It only takes one ruined DNA molecule to make a rogue cell. And
you dont require a carcinogen day after day to make a rogue molecule - it takes one chance event. One piece of bad luck amongst the billions of cell
your body contains. Cr6+ gives you lung cancer. The 5yr survival rate for that is 5% (eg see chemoleo's link). Its a death sentence, one that
generally takes 3-6 mnths to pass. You constantly sweat and feel like youre suffocating. Some argue there is no safe limit for a strong carcinogen
(multi-ring anthracenes, b-naphthalamine are such). Its not a case of you need such-and-such a dose of carcinogen before you can get cancer. One
small exposure can do it. Continual exposure just ups the probability.
[Edited on 19-3-2008 by len1]
|
|
|
woelen
Super Administrator
Posts: 8011
Registered: 20-8-2005
Location: Netherlands
Member Is Offline
Mood: interested
|
|
Indeed, one small exposure can do it. But isn't that true for all things. It remains a matter of probability. If I increase the chance of getting
cancer within the next 1 year from 0.01 to 0.01001, then I do not really care. E.g. I am strongly against smoking cigarettes. Each cigarette may
increase the chance by 0.00001 or so. But if you smoke 5 cigarettes each day, then the chance increases to considerable levels after some years of
smoking. But with New Years eve I usually smoke a few cigarettes in order to be able to light fireworks more easily. These few cigarettes increase the
chance and I could indeed have the bad luck that one of these triggers a starting cancer. But if you reason like that, then you hardly can live
anymore. Everything around us increases cancer risks, even the oxygen we breathe by means of radical formation. I look at these things in a relative
fashion. If a certain one-time behavior increases risk by 0.000...% then I don't care, as long as it remains one-time behavior. And this
peroxochromate experiment for me is such a one-time thing. Fun to play with for a few hours (spread over a few days) and then we move on to something
else. Most of the time I am not in my lab, and then most of the times, when I am in my lab, I am not working with carcinogens.
|
|
|
len1
National Hazard
Posts: 595
Registered: 1-3-2007
Member Is Offline
Mood: NZ 1 (goal) - Italy 1 (dive)
|
|
Yes I agree with all that. The big problem is that it depends on the numbers being what you say. Even a 1:1000 increase in that chance event, i.e.
going from 0.01 to 0.011 a 10% increase for instance is not acceptable for me. Especially as its entirely avoided with a gas mask. A gas mask adds
some certainty to the picture that what Im doing is not harming me - and that adds to the enjoyment. After all the idea is to have a good time right?
As regards everything having some carcigenicity - I think its a spectrum. Some things (vitamin C E) contribute negatively to the total probability of
cancer, some things (salt) have almost no effect, others (b-naphthelamine, benzylidene) have a huge positive contribution. Of what we know for
chromium is thst Cr6+ per day has a much greater positive contribution to cancer than the average number of cigaretes smoked per day by those plating
workers.
[Edited on 19-3-2008 by len1]
|
|
|
Fleaker
International Hazard
Posts: 1252
Registered: 19-6-2005
Member Is Offline
Mood: nucleophilic
|
|
While len has valid concerns, I am with Woelen and Chemoleo: small time exposures really don't add up worth a hill of beans. If one chance event to
cause a mutation is all that's needed, then why ever go outside into the sun, or drink chlorinated water or eat fried fish and chips? Also I wonder
about some individuals' genetic susceptibility to cancer: I've known people that smoked 60 cigarettes per diem for 50 years and didn't die from
cancer, or heart disease or an old (87) organic chemist who used to wash his hands with benzene and chlorinated hydrocarbons back in the 1950s. With
risk factors like that, you'd think they would be getting cancer, no?
I don't think a mild, one time exposure will have much effect, especially with Cr(VI), a comparatively mild carcinogen. At least so I'd like to
believe: I've used CrO3 on many occasions, and I've been working with dimethyl sulfate for the past couple weeks and I can tell you all what it smells
like :-/
I think I need a cigarette now :p
[Edited on 19-3-2008 by Fleaker]
Neither flask nor beaker.
"Kid, you don't even know just what you don't know. "
--The Dark Lord Sauron
|
|
|
Taoiseach
Hazard to Others
Posts: 241
Registered: 16-3-2008
Member Is Offline
Mood: No Mood
|
|
Peroxochromates
I'm still quite positive I do have the right stuff, simply because I have found a lot of references in literature which describe peroxychromates as
red-brown, yet not a single one describes them as black.
Nevertheless woelens hypothesis of a chromate-peroxychromate double salt sounds very plausible. I will test my compounds with BaCl2. I figure the
hypothetical double salt would dissociate into CrO8(3-) and CrO4(2-) in aequous solution, the latter would precipate as light yellow insoluble barium
chromate on addition of Ba(2+).
I'm very curious about the outcome because it could as well be the other way around! It is not unusual that mixed-valency compounds look black, so
woelens stuff might actually contain Cr(V) and Cr(VI). Possibly due to decomposition of the peroxychromate at temperatures close to 0°C, according to
the reaction
2[Cr(O2)4] (3-) + H2O ===> 2CrO4(2-) + 2OH(-) + 3,5O2
The difference between woelens and my synthesis is that I used much lower temperatures and (I think) less alkaline solutions.
There's a very interesting paper "EPR Spectroscopic Studies on the Formation of Chromium(V) Peroxo Complexes in the Reaction of Chromium(VI) with
Hydrogen Peroxide" which proves that there at least 2 more species are formed upon the addition of H2O2 to alkaline chromate solutions:
[Cr(O)(O2)2(OH2)] (-)
[Cr(O2)3(OH)] (2-)
The formation of [Cr(O2)4] (3-) is favored by excess of H2O2. It is actually proven to be formed over a wide range of pH if excess H2O2 is
present.
So could it be that woelens and my compounds differ by the number of peroxo-ligands, because they get partially replaced by oxo- and hydroxo-ligands
depending on temperature & pH?
Anyways we can already conclude from that paper that a strong excess of H2O2 is more important than high pH. A final writeup on the synthesis
should pay attention to that. I would rather use a huge excess of H2O2 and hamper decomposition of peroxychromate by very low temperatures
rather than highly alkaline solution.
Btw I have found a much better photo from my perchromate synthesis which shows the half-frozen mass just after the H2O2 has been added. You can see
the thermo isulation box (I made from styrofoam pieces) which was filled with 3 parts ice + 1 part table salt. This made sure the whole mix never
completely melted and temperature was well below 0°C all the time. Of course this could have been just the wrong decision and cause chromate to be
included in the crystal lattice... we will see!

|
|
|
Taoiseach
Hazard to Others
Posts: 241
Registered: 16-3-2008
Member Is Offline
Mood: No Mood
|
|
Peroxychromate impact test
A word about the impact test: It is actually not trivial to hit a sample of a very mobile powder with a hammer such that the bottom of the head smacks
flat against the anvil. If it does not hit flat, most of the energy will be directed to a single spot and dissipate. I remeber I hit my sample of
peroxychromate very hard with a medium-sized hammer and it did not react at all! It took me some time to realize that with such high momentum it was
almost impossible to let the hammer hit flat against the anvil. I finally succeded in blowing it up by wrapping the sample into aluminium foil and
hitting it with much less force, making sure the hammer hits flat and drives all the energy into the sample.
|
|
|
Taoiseach
Hazard to Others
Posts: 241
Registered: 16-3-2008
Member Is Offline
Mood: No Mood
|
|
diperoxotriamminechromium(IV)
woelen was right about diperoxotriamminechromium(IV); its forumla is [Cr(NH3)3(O2)2] and not [Cr(NH3)3(O2)4].
There is a synthesis for this compound in Brauer's Handbook of Preparative Inorganic Chemistry, vol 3.:
Cool a mixture of 25 ml 10% ammonia and 5ml 50% solution of CrO3 to 0°C, then add 5ml 30% H2O2. Let is stand in an ice bath for 1h. Then warm
to 50°C until almost all has redissolved and the vigorous bubbling has ceased. Filter to remove precipated (NH4)2Cr2O7 and cool the clear liquid to
0°C. Yield: 0,3g.
(Can anyone compute the right amount of ammonium chromate from these details? I have no clue how much CrO3 there is in 5ml of a 50% solution)
Properties: Light-brown needles, soluble in dilute ammonia, slightly soluble in water under decomposition, insoluble in other solvents. Explodes on
heating.
I start to believe that my putative [Cr(NH3)3(O2)2] is actually not a pure compound but a mixture of perchromate and the ammine complex. It
does contain light-brown needles, but also some other stuff more dark in color. I did not warm the solution to 50°C when I made it, so that
might be the reason not all perchromate was converted.
To add to the confusion, there's a paper "Some Decomposition Reactions of diperoxotriamminechromium(IV)" by R.G. HUGHES, E.A.V. EBSWORTH and
C.S.GARNER. It describes the compound as lustrous black needles. I once obtained such black needles from K2Cr2O7 and H2O2 in
ammoniacal solution at room temperature. However, when I tried the experiment again, I obtaind a mixture of black needles and fine red crystals. It
might have been a change in temperature and/or concentration of ammonia which caused the different outcome. The red stuff is said to decompose in
ammonia water in the K. A. Hofmann und H. Hiendlmaier paper. In their diperoxotriamminechromium(IV) syntheses, they always produce a mixture of
peroxychromate and the ammine complex, and then wash away the peroxychromate with ammonia water at room temperature. At least, this is consistent with
Brauer's synthesis.
The original paper on peroxychromates by K. A. Hofmann und H. Hiendlmaier says that the reaction depends on both temperature and concentration of NH3
and either yields ammonium perchromate or the triammine complex. However, that paper dates back to 1905. There is another statement from ERNSTH .
RIESENFELD, WILLIAAM. KUTSCH and HERMAN OHL which is of great interest: "Contrary to Hofmann and Hiendlmaier’s statement (Abstr., 1905,ii, 716),
the action of hydrogen peroxide on ammonium chromate, whether
containing 2.5 per cent. of free ammonia or saturated with ammonia,leads to the formation of ammonium perchroinnte, (NH,),CrOs, unless insufficient
peroxide is used or the temperature is allowed to rise above 0°C, when a mixture of ammonium perchromate and Weide’s triaminine chromium tetroxide.
Cr04 3NH3 (Abstr., 1898, ii, as), or the latter alone, is obtained. When treated with 10 per cent. ammonia, ammonium perchromate changes immediately
at 40°, but
only slowly at the laboratory temperature, into Weide’s compound, which separates in needles and rectangular and rhombic plates. The three forms
have the sp. gr. 1.964 at 1 5-S0, and when examined crystallographically appear probably to be identical, and do not constitute two isomeric
substances as stated by Hofmann and Hiendlmaier (Zoc.
c i t . ) . The action of dilute sulphuric acid on Weide’s cornponnd leads to the formation of chromium sulphate and hydrogen peroxide, but not of
chromic acid ; the amount of oxygen evolved varies from 3.54 equivalents with 1 per cent. to 2.24 equivalents with 20 per cent.
acid."
Perfect confusion then... 
To make things even worse, the R.G. HUGHES, E.A.V. EBSWORTH and C.S.GARNER paper indicates that the ammine-complex forms NH3 upon addition of NaOH and
warming, so that might not be a good way to distunguish it from ammonium perchromate. Damn
[Edited on 21-3-2008 by Taoiseach]
|
|
|
Taoiseach
Hazard to Others
Posts: 241
Registered: 16-3-2008
Member Is Offline
Mood: No Mood
|
|
Btw check this out:
CALCIUM PERCHROMATE. A NEW TYPE OF RED
PERCHROMATE
Its the first google hit that turns up. Full pdf can be downloaded FOR FREE.
It describes that addition of H2O2 to freezing cold chromium hydroxide leads to formation of what they call "red perchromic acid". Addition of calcium
chromate or acetate leads to the formation of calcium peroxychromate. I think they are wrong about structural formula and valency tough. The paper is
from the early 30s so at that time they might not have fully understand the complex coordinative chemistry behind peroxo-chromium compounds.
Anyways this compound might be useful to prepare other perchromates by double displacement reaction with metal sulfates. Also, barium
peroxychromate could be prepared conveniently from this "red perchromic acid" as well.
One might even dream about nickel hydrazine peroxychromate - a compound right out of devil's kitchen
I have papers on Rb and Cs peroxychromates (including synthesis), but they are worthless for practical experiments. The Cs compound explodes upon
seperation from the solution and the Rb compound is unstable at room temperature.
[Edited on 21-3-2008 by Taoiseach]
|
|
|
woelen
Super Administrator
Posts: 8011
Registered: 20-8-2005
Location: Netherlands
Member Is Offline
Mood: interested
|
|
I made a webpage about the triammine peroxo complex of chromium(IV):
http://woelen.homescience.net/science/chem/exps/Cr_ammine_pe...
This compound has properties, close to those, described by Taoiseach. It burns very fast, when heated.
Next project will be the making of the "red perchromic acid". Just making Cr(OH)3, filtering/rinsing and adding H2O2 does not sound very difficult.
The element chromium, combined with peroxide, is good for MANY interesting experiments.
Whoever tries to repeat this experiment: be VERY careful, avoid inhaling of fine droplets of the fizzling solution, and do not scale up. I am not sure
about the stability, and if it deflagrates, then the damage from 100 mg can be overcome, but the damage of 10 grams may be a disaster!! You are
warned.
The picture below shows my K3CrO8 and the Cr(NH3)3(O2)2:

Edit(woelen): Made links work again
[Edited on 6-4-15 by woelen]
|
|
|
woelen
Super Administrator
Posts: 8011
Registered: 20-8-2005
Location: Netherlands
Member Is Offline
Mood: interested
|
|
I have done some more reading on the subject as well. I have another book, "Inorganic Chemistry, a text-book for advanced students" by Barry, Barnett
and Wilson. This book covers the peroxo chromates. It describes the "red peroxochromates" on page 471. These are dark red/brown compounds. According
to this book, the potassium salt is a stable salt, which easily is prepared by adding 30% hydrogen peroxide to an alkaline solution of potassium
chromate below 0 C. According to this book, the sodium salt and ammonium salt cannot be prepared in a dry state.
The pure potassium salt is stable, but it explodes at 178 C when it is heated. The wet sodium salt and wet ammonium salt have been prepared, the wet
sodium salt explodes above 115 C, the wet ammonium salt explodes above 50 C.
Remarkably, this book assigns another structure to the red peroxo chromates as what I have done up to now. The ion is said to be Cr2O16(6-), it is a
chromium(VI) compound, and it has the following structure:
(-OO)3Cr(=O)OOCr(=O)(OO-)3
So, there are 7 peroxo groups in this ion, and two oxo groups.
The book also mentions a neutral triammine complex on page 478, which also is described as a chromium(VI) compound, and its structure is
Cr(O2)O2(NH3)3. It contains two oxo ligands, one peroxo ligand and three ammine ligands. This is the compound we have made, we, however, regard it as
a chromium(IV) compound with two peroxo ligands.
Another peroxo chromate is mentioned, also a chromium(VI) compound, and this can be made by adding ice cold hydrogen peroxide to a solution of
potassium dichromate, without any acid or alkali added. Crystals of a dark blue compound K2Cr2O12 are formed. This is something I'll try next week at
a test tube scale.
The descriptions of the physical properties and methods of preparation (or impossibility thereof) are very valuable. The structures, given in this
book, however, must be taken with a grain of salt. This is a fairly old book from 1960. But again, more confusion is added.
|
|
|
| Pages:
1
2
3
4 |
|