| Pages:
1
..
19
20
21
22
23
..
33 |
nitro-genes
International Hazard

Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
It's not due to a colour shift from changes in pH, dinitro N-nitroso acetaminophenol is an existing compound (CAS RN: 146897-61-2 ), stability and
toxicity unknown.  the difficulty is determining what percentage of the dark
brown impurities are from remaining tablet binders versus from nitration, all I know is that the amount of impurities filtered out varies per batch of
acetaminophen/nitration, usually though it represents only a minor fraction compared to the iso picramic acid obtained. the difficulty is determining what percentage of the dark
brown impurities are from remaining tablet binders versus from nitration, all I know is that the amount of impurities filtered out varies per batch of
acetaminophen/nitration, usually though it represents only a minor fraction compared to the iso picramic acid obtained.
No luck yet with DDNR, an attempt to nitrate the isopicramic acid acid further to the putative trinitro nitramine compound (0 to - 20 deg C, 24 hours,
AN/SA, pouring on ice) resulted in a lot of foaming and precipitation of a bright yellow compound with definite smell of p-benzoquinone.
Recrystallization/hydrolyzation from refluxing in 96% ethanol for 30 minutes , followed by addition of a potassium acetate solution at pH 6 and
cooling seemed to precipitate only p-DDNP. Also no luck with oxidative nitration of pDDNP at elevated temperatures, or boiling with excess nitrite.
Searching for optimal reaction conditions and subsequent purification without a way to monitor tthe reaction at various stages makes it next to
impossible to come to a conveniant route for DDNR IMO.
There are three experiments I would still like to perform, any thoughts on the outcomes of these reactions would be much appreciated:
1. Add 3 grams of the putative dinitro N-nitroso acetaminophenol to an acidified (equimolar or slight excess) H2O2 solution
2. Add 3 grams to 65% nitric acid letting it stir at room temperature, or 60 deg C while monitoring colour changes.
3. Adding the putative 3 nitro acetaminophen from nitrite nitration to 65% nitric acid at 0 deg C for 12-24 hours.
[Edited on 16-8-2015 by nitro-genes]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Usually the boiling of a diazo compound with alcohol will eliminate the diazo and leave the unsubstituted hydrogen ring where the diazo is removed.
That is the "known reaction with alcohol" that is mentioned in the literature.
I don't think it will be possible to add two nitros to isopicramic acid to reach a trinitro N-nitramine when even adding 1 nitro is a push, and the
trinitroaminophenol is only tenuously and marginally, transiently stable, with that third nitro at position 3 prone to decomposition to a hydroxyl,
which is okay since that will lead to DDNR.
I'm not clearly understanding what you are trying to do.
[1] From where are you obtaining the supposed N-nitroso acetaminophenol and what are you trying to convert it to using H2O2? I don't see this likely
to lead to any diazo compound. I'm not sure there even is an N-nitroso acetaminophenol that is what you have.
[2] ditto
[3] Are you talking about the 3-nitroso acetaminophen via Masunto and not 3-nitroacetaminophen? I don't know of any nitrite nitration that produces
the 3-nitroacetaminophen but only the 3-nitroso or the 2-nitro. The only 3-nitroacetaminophen was described gotten using the molybdenum catalyst with
nitric acid, or by using the acetaminophen acetate as a starting material for the nitration.
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
My bad, by refluxing in ethanol I meant keeping the solution at 40 deg C. (Boiling point of DCM), which is not refluxing indeed and unlike DCM a polar
solvent. I did not aim for the tetranitro compound, but was hoping that possible
formation of the primary aryl nitramine at very low temperatures to avoid decomposition would lead to intramoleclar rearrangement of the nitramine to
produce either the trinitro diazo or lead to hydrolysis of the 3 nitro group to produce DDNR, both described and existing compounds. I recognize these
routes are not the most hypothetically likely to succeed, though unlike the described non-OTC routes from literature to trinitroacetaminophen and
DDNR, I'm simply reasoning from an simplicity and OTC standpoint for which either isopicramic acid or DNAc as a possible reaonably storage stable 1 or
2 step precursor to both p-DDNP and DDNR is worth investigating using small scale experiments. Another, but more complicated, OTC route would involve
O-protection of acetaminophenol by reaction with benzylchloride as described by Meldola (which is OTC produced from benzylalcohol, perfume ingredient
found online and concentrated HCl, (orgsynth)) and nitration using only NA.
The thing I had in mind for the first reaction is nitrating acetaminophenol to DNAc using NA, crashing the nitration mix on ice and adding a solution
of one mole equivalent of nitrite to produce the bright orange putative N-nitroso dinitroacetaminophenol then try oxidation using either hydrogen
peroxide or NA to obtain trinitro acetaminophenol via rearangement of the putative nitramine or lead to a decomposition leading to deacetylation, both
options would be fine by me. The molybdenum catalyst seems interesting as well, saw it posted in the reference section and would be interesting to
verify. As a gashead I'm thinking MoS2 + O2 --> MoO3 + NH3 --> (NH4)6Mo7O24 * 4 H20 -- > 100 deg C. --> (NH4)6Mo7O24 
[Edited on 17-8-2015 by nitro-genes]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Who would have thought
It seems isopicramic acid can be conveniently and completely OTC diazotized by treating AN/SA/H2O with fine pieces of metallic copper from electricity
wire. The procedure probably works to produce very pure pDDNP since nitrous gasses (NO and NO2) are formed in situ, whereas the reaction neither
produces a significant amount of H2, nor is copper (unlike iron, zinc, aluminium etc) able to reduce the nitrogroups. Still needs some optimization,
though it definitely works, and seems to produce better yields than heating in 65% nitric acid alone.
I used something like this:
1 gram of finely powdered isopicramic acid was suspended in 8 ml of water. Then 3 ml's of 96% SA was added and the suspension heated with stirring to
produce a solution of isopicramic sulfate. When cooled to 30 deg C., 4 grams of ammonium nitrate was added and 1 gram of metallic copper. The mix was
kept at 50 deg C for 1 hour and crashed on ice to precipitate 0.8 grams of pure p-DDNP.
|
|
|
greenlight
National Hazard
Posts: 734
Registered: 3-11-2014
Member Is Offline
Mood: Energetic
|
|
Nitro-genes, how big were the pieces of copper wire or was it one piece. Do you think copper powder could be used instead of wire and filtered out
before crashing into ice.
Be good, otherwise be good at it
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
The copper came from 2 mm thick solid eleectricity wire, cut with siccors to about 3 mm long "beads" that can be stirred with the magnetic stirrer bar
without doing too much damage to the teflon coating. Be sure you strip the wire of any potential protective coatings else it will contaminate your
product. The procedure is not optimized yet, it was more of a curiosity for me, so it may still need some tweaking.
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
That is a clever variation for the diazotization scheme using copper to generate the NO2. Was that a method you copied from somewhere or was that a
brain storm of your own invention? Reduced copper powder from copper sulfate and vitamin C would probably be more active and produce a higher yield
more quickly.
precipitated copper powder has a discussion thread here
http://www.sciencemadness.org/talk/viewthread.php?tid=2654
[Edited on 8/25/2015 by Rosco Bodine]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Thanks, and no, I thought of it myself, reasoning that most metals are able to reduce nitric acid (wheres organic reductors tend to give a mess), I
reckoned needing an OTC metal that did not react directly with H30+ such as more stronger reducing metals as zinc or iron, and did not produce any
precipitate.
You bring up an interesting question though: What is the primary nitrogenoxide species that leads to formation of the diazogroup? I was under the
impression that NO (being the anhydrate of nitrous acid) is. This may be important, since depending on the dilution of NA, it produces primarily NO or
NO2 from the reaction with metallic copper. And you are right, the copperpowder would react much faster (maybe resulting in less by-products), on the
other hand, the exotherm may be more diffult to control and it would need carefully weighed amounts to be sure no copper powder is left, whereas the
beads can simply be filtered out and used again.
The really funny part is that the method described above, using metallic copper and nitrate salts/ sulfuric acid, produces the most free flowing, high
density pDDNP I've seen till so far. Maybe the addition of the sulfuric, high
temperatures or the presence of metallic copper or it salts seems to somehow affect crystal formation to what it seems, sort of rounded shapes.
[Edited on 25-8-2015 by nitro-genes]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
You are onto something good there with the use of copper in wire form.
The fine copper wire could have its advantages by the very gradual reaction which results in a very slow diazotization which is similar to the use of
nitrous fumes being bubbled from a gas dispersion tube, but is occurring in situ with the extremely fine bubbles coming off the copper wire. For
finer copper wire the wires from stranded copper wire like lamp cord would be good and even finer copper wire strands are found in old vacuum cleaner
cords. The copper braid that is found in coaxial cable is fine wire also but is woven into a sleeve which might need to be unraveled and teased apart
into loose pieces.
The dissolved spent copper in the diluted solution could be recovered later for other uses as low soluble basic copper carbonate. A rough
neutralization first done using lime to precipitate the sulfate value could leave mostly copper nitrate as an alternative material and useful
byproduct itself, or which could be converted to the energetic tetraamine copper nitrate using ammonia. Waste not want not.
As to whether it is NO or NO2 that accomplishes the diazotization maybe it is both as N2O3  and it is reported that way sometimes and has been reported that NO alone can work or NO2 so it is ambiguous. It may be that exposure
to the air oxidizes the NO to NO2 which then does the deed. and it is reported that way sometimes and has been reported that NO alone can work or NO2 so it is ambiguous. It may be that exposure
to the air oxidizes the NO to NO2 which then does the deed.
This could account for the use of formaldehyde being possible to cause a diazotization by its reducing effect on HNO3 which leaves no residue
byproduct, as occurs for some other reducing agents like starch used with HNO3 in a gas generator.
The reaction of copper with nitric acid is probably
Cu + 4HNO3 -----> Cu(NO3)2 + 2NO2(g) + 2H2O
Anyway the use of copper wire in situ in a diazotization scheme is a new one on me and it is a very good idea.
The nascent nitrous fumes are also a highly active nitrating agent even in dilute acid solutions, where something like salicylic acid sulfonate is
nitrated all the way to picric acid under mild conditions, and in quantitative yield, so this use of copper could also have other very interesting
uses. Excellent.
[Edited on 8/26/2015 by Rosco Bodine]
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
Here are some references from Nicodem:
4-amino-2,6-dinitrophenol and 4-diazo-2,6-dinitrophenol
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
3-diazo-picric acid (exist?) possible? precursor for iso-DDNR (exist?)
If they don't exist yet, then let us invent them here
In a couple of posts last month by nitro-genes has been provided a couple of pieces of very interesting information, regarding reaction of picric acid
with hydroxylamine to form 3-amino picric acid. It is a synthesis where the lithium salt of picric acid was used for the increased solubility. I have
speculated magnesium picrate may also be workable and more economical.
http://www.sciencemadness.org/talk/viewthread.php?tid=62326&...
and an easy preparation of the needed hydroxylamine
http://www.sciencemadness.org/talk/viewthread.php?tid=439&am...
In some U2U messages I have shared an idea about this on which our thinking may be parallel.
3-amino-2,4,6-trinitrophenol is isomeric with the 4-amino-2,3,6-trinitrophenol of Meldola and also Klapotke which can be diazotized to form a
diazotrinitrophenol having marginal stability which has a nitro that decomposes to a hydroxyl and forms DDNR.
It may be that similarly the 3-amino picric acid will likewise be possible to diaztotize to form a marginally stable diazotrinitrophenol which
likewise is subject to decomposition of one of its nitro groups to form an isomer of DDNR.
Neither of these hypothesized energetic diazo compounds have so far turned up in any search of the literature
Here is the 3-amino picric acid which would be the precursor

Attachment: US8404897 3-amino picric acid.pdf (550kB)
This file has been downloaded 665 times
[Edited on 8/27/2015 by Rosco Bodine]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
The main thing I had in mind for the 3-amininopicric acid was KNDB, a mil spec approved replacement for LS, which is formed from azido reduction of
3-azido-2,4,6 trinitrophenol with K2CO3 at room temperature. I don't think it is a viable precursor to DDNR. The synthesis is listed in:
US 8748639 B1
3-azido-2,4,6-trinitrophenol, method of making, and method of transforming
"Example 1
12 mL of concentrated sulfuric acid were charged to the flask depicted in the experimental setup, and agitation was initiated. 0.897 grams (0.0037
mol) of 3-APA was then added and rinsed down with an additional 3.5 mL of concentrated sulfuric acid. The 3-APA dissolved readily and heating the
reaction mixture was unnecessary. The resultant solution was cooled to 3° C. using an ice water bath, and 0.39 grams (0.0057 mol) of NaNO2 dissolved
in 6 grams of concentrated sulfuric acid was added dropwise, keeping the temperature below 5° C. This addition took less than 5 minutes. The
resultant solution was then stirred for 20 minutes at that temperature. 25 mL of phosphoric acid was then added at such a rate that the temperature
did not exceed 5° C. This addition was somewhat exothermic and occurred over 45-50 minutes. After addition was complete, the reaction mixture was
stirred at less than 5° C. for 10 minutes. During the addition and subsequent stirring, the reaction mixture became quite viscous and attained a
pink-orange color. 1.18 grams (0.018 mol) of NaN3 was then added in portions, keeping the temperature below 5° C. This addition took less than 5
minutes. The reaction mixture was then stirred at less than 5° C. for 20 minutes, and was then allowed to come to room temperature by removing the
ice water bath. Once the reaction mixture had reached room temperature, it was poured into 70 mL of cold (<5° C.) water with stirring. The
resultant yellow precipitate was then collected by filtration, and washed with 2×20 mL of cold water. The yellow solid exhibited some water
solubility, so the water washed were retained for further analysis and/or crystal growth. The remaining precipitate was then dried in a dessicator,
and yielded 0.44 grams (44% yield)."
Looks like a very dangerous synthesis adding NaN3 to icecold SA, definitely not suitable for scaling up. Not sure what the phosphoric acid addition is
for, presumably diluting the hN3 formed, and why no urea is added after the nitrite step. Anyway, since no precipitation is mentioned and a low
temperature is used, it seems logic that the diazo derivative is presumably not very stable. My main interest was (after experimenting with the
tetrazeno derivatives, which may just be an azido), that this synthesis might be performed with hydrazine sulfate instead. A two option experiment,
where either the azido or tetrazeno derivative may form both of potential interest.
Another experiment I had in mind (though not very likely) is oxidative nitration of 3-aminopicric acid using 65% NA and moderate heating, which might
produce the furoxan directly (can't imagine that it would have been found earlier, though very patentable if it is ). Results most likely in either picric acid or stypnic acid, though worth a shot
maybe. Anyway, more ideas than time than usual...
VNS reaction of picric acid and hydroxylamine is performed at extremely high pH by addition of lithium hydroxide. Was there a particul reason you
mentioned the magnesium salt? Wouldn't all magnesium picrate instaneously precipitate as magnesium hydroxide? Unlike zinc and aluminium, I'm not
aware of a magnesiate being formed from dissolution in NaOH.
the N-O-diacetaminophen nitration to trinitroacetaminophen also brought me another idea. Don't know theoretical background on the most likely ring
substitutions, but what if benzoxazolone was nitrated in 65% nitric acid? The N position may be more dominant in oriantating nitro groups orho para,
but the alkylated O is more suscpetable to hydrolysis, which shifts the orientation during the reaction, likely very similar to the formation of
trinitroacetaminophen klapotkes group described. Curious what the outcome may be.
Unlike trinitroacetaminophen, which needs Ac2O or very unhealty alkylation agents, benzoxazolone is potentially completely OTC from salicylic acid
--> salicylamide --> treatment with household bleach (and maybe sulfide to prevent ring chlorination).
[Edited on 27-8-2015 by nitro-genes]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Regarding US8748639 (attached)
It looks like what they are doing is forming the soluble diazonium acid salt and then displacing the diazo with an azide to form another interesting
compound.
However if the soluble diazonium was simply diluted, then the diazo oxide would likely precipitate. The diazo at position 3 would form a conjugate
linkage with the hydroxyl at 1, and hydrogen would be displaced to form the anhydride or "diazo-oxide".
They show a chlorine there which is an interloper from somewhere that should not be there

It may be that the arrangement of the three nitro groups is stable and that there would be no decomposition during an extended digestion as may lead
to an isomer of DDNR. For this possible decomposition the diazo oxide would be heated with potassium acetate solution to form the potassium salt of
an iso-DDNR.
In the scheme I was contemplating there would be no displacement using azide of the diazo intermediate, but the diazo would be left intact.
What I am contemplating is a structurally analogous scenario which is possible that solves the futility of trying to introduce a third nitro at
position 3 on isopicramic acid. The general reactions which involve diazotization and subsequent hydrolysis of one of the nitro groups of the
resulting diazotrinitrophenol (anhydride) are believed might occur the same, leading to an iso-DDNR.
Attachment: US8748639 3-azido-2,4,6 trinitrophenol.pdf (469kB)
This file has been downloaded 881 times
In regards to the US8404897 process and use of magnesium being potentially a problem that is possible and I have not even worked out the stoichiometry
to look at the molar equivalents but had expected the reaction would proceed at a pH where the magnesium would not precipitate. Anyway you had said
the reaction does proceed using sodium so that already provides a cheap alternative to the lithium if magnesium is unworkable. Magnesium was just an
idea that seemed like a candidate if the pH requirement is not extreme. Ammonia might be helpful along with the magnesium and I don't recall for sure
but IIRC ammonia and ammonium nitrate may work together to enhance solubility the same way as works for zinc if the high pH is required. I'll have to
look that one up. If lithium is required to do things right, then lithium is required.
Update: I looked at the stoichiometry and see the reaction pH is very high, really caustic conditions so the use of lithium is pretty much required
for the good yields of 3-aminopicric acid reported by the patent.
Quote: Originally posted by nitro-genes  |
Another experiment I had in mind (though not very likely) is oxidative nitration of 3-aminopicric acid using 65% NA and moderate heating, which might
produce the furoxan directly (can't imagine that it would have been found earlier, though very patentable if it is ). Results most likely in either picric acid or stypnic acid, though worth a shot
maybe. Anyway, more ideas than time than usual...
|
I wouldn't worry about the furoxan, just dissolve the 3-aminopicric acid in HNO3 and throw in your copper wire .....ummmm, whatever works is good.
[Edited on 8/28/2015 by Rosco Bodine]
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
| Quote: Originally posted by nitro-genes | Who would have thought
It seems isopicramic acid can be conveniently and completely OTC diazotized by treating AN/SA/H2O with fine pieces of metallic copper from electricity
wire. The procedure probably works to produce very pure pDDNP since nitrous gasses (NO and NO2) are formed in situ, whereas the reaction neither
produces a significant amount of H2, nor is copper (unlike iron, zinc, aluminium etc) able to reduce the nitrogroups. Still needs some optimization,
though it definitely works, and seems to produce better yields than heating in 65% nitric acid alone.
I used something like this:
1 gram of finely powdered isopicramic acid was suspended in 8 ml of water. Then 3 ml's of 96% SA was added and the suspension heated with stirring to
produce a solution of isopicramic sulfate. When cooled to 30 deg C., 4 grams of ammonium nitrate was added and 1 gram of metallic copper. The mix was
kept at 50 deg C for 1 hour and crashed on ice to precipitate 0.8 grams of pure p-DDNP. |
Nice one!
Beware that Cu also reduces H2SO4 to SO2...what may reduce some nitrogroups...
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Thanks, was surprised it worked so well actually, since supposedly o-DDNP is destroyed by hot concentrated sulphuric acid, so it needs some water at
(least <60%) to prevent decomposition. On the other hand, very low sulfuric acid/nitrate salt concentrations may form mostly NO instead of NO/NO2
from the reaction with metallic copper and need stonger heating for the reaction to proceed within a couple of hours. With some tweaking, an optimum
can probably be found using sulfuric acid. It's pretty cool though, p-DDNP from acetaminophen, SA, ammonium nitrate and ammonia
Didn't smell any SO2, I reckoned the nitric acid oxidation would be much faster. May, at these temperatures, the SO2 even be oxidized by nitric acid
back to sulfate? In that case it may act like a catalyst for nitrous gas formation.
With only nitric acid/copper the reaction may even work with something like 15 ml of 20-25% nitric acid, which would be much more economical in terms
of nitric acid usage than using 65% and may even rival the use of nitrite in terms of costs. Unlike the diazonium chloride and nitrate (if it is
that), the diazonium sulfate is almost completely insoluble, thus may provde better control over crystal formation.
On the other hand, the decreased solubility of p-DDNP in the SA/nitrate salt reaction mixture may actually be the reason for the higher yield than
from nitric alone.
[Edited on 29-8-2015 by nitro-genes]
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
Since it was already spoken about it in this tread and that it comes back here, I reply here:
| Quote: Originally posted by PHILOU Zrealone |
posted on 20-5-2015 at 21:21
I wonder if 3-diazonium-2,4,6-trinitrophenol does exist...thus a DDNP variant with the diazo in meta of the phenol and with 3 nitros... thus based on
meta-amino-phenol.
I also have good synthetic ideas about 3,5-diazido-2,4,6-trinitrophenol.
Depending on the stability of iodoxy group vs phenol, nitro and diazonium group...one may also invesigate 3,5-diiodoxy-DDNP and 3,5-diiodoxy-TNP.
|
| Quote: Originally posted by nitro-genes |
posted on 21-5-2015 at 11:02
@Philou: 3-diazonium, 2,4,6 trinitrophenol must exist, since it is used as an intermediate in the synthesis of KDNBF, mentioned earlier. I've only
seen it used to prepare the azido compund and uses very strong diazotising conditions at low temperatures, I reckon it would't be very stable.
What was your idea about the synthesis of 3,5-diazido-2,4,6-trinitrophenol, sounds interesting
|
So here is my synthetic pathway to diazido-trinitrophenol!
Image:
Synthetic pathway to 3,5-diazido-2,4,6-trinitrophenol and hydroxy-azido-dinitrobenzofuroxan derivatives (5-azido-4,6-dinitro-7-hydroxy-benzofuroxane
(o-HADNBF - ortho-hydroxy-azido-dinitrobenzofuroxane); 7-azido-4,6-dinitro-5-hydroxy-benzofuroxane (p-HADNBF - para-hydroxy-azido-dinitrobenzofuroxane
); 7-nitro-8-hydroxy-benzo-di-furoxane (o,p-HNBDF - ortho-para-hydroxy-nitrobenzodifuroxane))
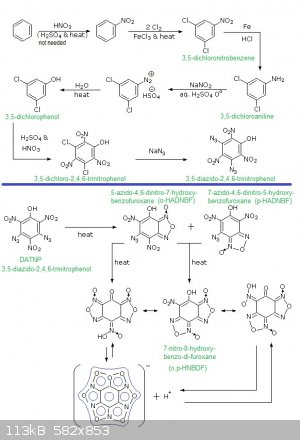
Remarks:
1°)Note that the process will also work for mono-azido-TNP via m-chlorophenol...
2°)I thought about it since the preparation of 1,3,5-triazido-2,4,6-trinitrobenzene (TAZTNB) is easy from 1,3,5-trichloro-2,4,6-trinitrobenzene
(TCTNB) and NaN3 in an appropriate solvent.
3°)Interesting fact: upon heating TAZTNB turns into hexanitrosobenzene (less sensitive and less powerful but stil as good as tetryl) what is in fact
a benzo-tri-furoxan...this change occurs at moderate speed at ambiant T° (few % over several years) but becomes significant at each 10°C T°
increase...
4°)Also a very good finding (for Rosco Bodine) is that TAZTNB is a good way to produce both azide salts and 1,3,5-trihydroxy-2,4,6-trinitrobenzene
(trinitrophloroglucidol)
1,3,5-trichlorobenzene -HNO3/Oleum-> 1,3,5-trichloro-2,4,6-trinitrobenzene
1,3,5-trichloro-2,4,6-trinitrobenzene + hydrazine --> 1,3,5-trihydrazinium-2,4,6-trinitrobenzene tri-hydrochloride
1,3,5-trihydrazinium-2,4,6-trinitrobenzene tri-hydrochloride + NaNO2 --> NaCl + 1,3,5-triazido-2,4,6-trinitrobenzene + H2O
1,3,5-triazido-2,4,6-trinitrobenzene + NaOH --> NaN3 + 1,3,5-trihydroxy-2,4,6-trinitrobenzene
5°)About 3,5-diazido-2,4,6-trinitrophenol and hydroxy-azido-dinitrobenzofuroxan derivatives exposed above: salts of heavy metals or of amines (NH3,
NH2OH, N2H4, (NH2)2C=NH, (NH2-C=NH)2), NH2-Tetrazole, NH2-NH-Tetrazole) must be quite powerful
[Edited on 1-9-2015 by PHILOU Zrealone]
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
possible alternative synthesis for o-DDNP
I have an idea for a possible alternative method of synthesis for o-DDNP based upon nitration of salicylic acid to 3,5-dinitrosalicylic acid,
followed by reduction by sugar to 3-amino-5-nitrosalicylic acid. This would be subjected to a combined diazotization and further nitration with
accompanying decarboxylation to form o-DDNP.
Aspirin could be used I think as the starting material since it would be easily deacetylated by the nitration mixture leading to 3,5-dinitrosalicylic
acid. The sodium salt would be formed and dissolved in hot water for reaction with fructose or glucose.
It is unknown what is the solubility of the 3-amino-5-nitrosalicylic acid. It may be necessary to first diazotize and precipitate the
3-diazo-5-nitrosalicylic acid as an intermediate and then complete the nitration and decarboxylation to form the o-DDNP. It is expected that this
final step will occur under relatively moderate conditions. The solubilities and conditions for manipulations will affect the process and may even
determine if the process is workable or not. But the reactions work in theory and could provide an easier route to o-DDNP which avoids picric acid.
Attachment: Dinitrosalicylic_acid_as_a_reagent_for_the_estimation_of_sugar.pdf (344kB)
This file has been downloaded 947 times
Attachment: Meldoa et al 5-Aminosalicylic acid and derivatives 533.pdf (326kB)
This file has been downloaded 903 times
Meldola et al obtained what is evidently the material which is 6-carboxyl potential precursor for o-DDNP ( 3-diazo-5-nitrosalicylic acid ) via
diazotization of 3-amino-5-nitrosalicylic acid using HCl and sodium nitrite. Those conditions would leave the carboxyl intact. If that 6-carboxyl
precursor for o-DDNP so obtained by Meldola was simply dissolved in HNO3, it is anticipated the carboxyl would be eliminated and substituted by a
nitro, to produce o-DDNP.
What I anticipate would occur if the diazotization was performed under alternative conditions as would use HNO3 instead of HCl, or possibly to use the
method of Benedikt and Hubl for the diazotization, the carboxyl would be eliminated and a nitro introduced in its place, and the result would be
o-DDNP.
One or all three of these anticipated reaction schemes should produce o-DDNP as the end product.
As a related experiment it might be interesting to see what may result from a nitration of Molybdenum Salicylate, or possibly Chromium Salicylate to
see if such a scheme might via an influence of the metal directing the first entering nitro group to a different ring position lead to a different
dinitrosalicylic acid, or a trinitrosalicylic acid, or a trinitrophenol, or possibly even a tetranitrophenol.
A search of the literature so far is not returning information of any use in further documenting these ideas.
[Edited on 9/1/2015 by Rosco Bodine]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Hi Philou, thanks for this synthesis scheme, some interesting compounds, some of them never described before, especially the o,p HNBDF looks like a
very interesting molecule, do you think it would be able to form salts? The synthesis route described looks doable, and apart from the NaN3 (which can
be made OTC though) completely OTC. Pretty cool!
Some things I wondered about :
1 How selective would the chlorination of nitrobenzene be in producing only 3,5 dichloro nitrobenzene? This can probably be found in literature, but I
haven't had the time yet to get a good look.
2. How selective is the Fe/HCl reduction in only reacting with the nitro group? Is there completely no dehalogenation? Does it need a certain strenght
of HCl, or are acidic conditions enough? I'm asking, because probably, mono and dichloro aniline would be hard to separate
3. What would be the best solvent for the diazotization step, since dichloroaniline is not soluble in water, though it seems to be in ethanol. Ethanol
can be used in some cases, but can give some coupling products IIRC.
4. It would be really cool to see how easily the resulting 3,5 dichlorophenol would be able to nitrate further as opposed to 1,3,5 trichlorobenzene.
Do you have a reference for this nitration? Curious about how stable the chloro groups would be for the phenol.
5. the 3,5 dichloro 2,4,6 dinitrophenol would be a very nice precursor to have, the reaction with ethanolic hydrazine may produce a Di N-hydroxy
benzotriazole, which has never been described before I think. If it would be stable that is, N-hydroxy benzotriazoles are notorious for explosions
during synthesis IIRC. Definitely a nice mad science project, nice!
@ At Rosco, haha amazing, had the same idea and wanted to try it once! I never
got around to finding a proper synthesis protocol for dinitrosalicylic acid, so thanks for the references. Also wondered how much easier the dinitro
salicylic acid would reduce one nitro group, as opposed to trinitrophenol. Every TNP reduction scheme, other than with either commercial or homemade
hydrogensulfide, seems to result in an mess from which only small amounts of picramic acid can be isolated. But indeed, this may be worth a try, I
read the first reference you provided, and it indeed seems more selective than picric acid. There can be a big difference though between umol
quantities as quantitative test and a high yield synthesis. Same goes for ascorbic acid and picric acid, which seems to form some form of inseparable
complex with either picric acid or picramic, I read an article on this, but can't find it again, sorry.
Another question for those who may know more about organic chemistry than me:
Benzoxazolone can be made in high yield, completely OTC. From literature it seems the 6 and 5 positions are most activated, and 6-nitro benzoxazolone
can be made from just 65% HNO3 in high yield by nitration at 0 deg C. Now, could you hydrolyze the oxazolone ring using reflux conditions and dilute
mineral acids, or would the nitro group decompose? If you would get 2-amino 5-nitrophenol and nitrate it further, what would be the outcome? I
couldn't find anything on this, not even for 3-nitrophenol. Suggestions for followup or stability of these compounds? This could be really
interesting, as most of the reactions involved are smooth sailing.
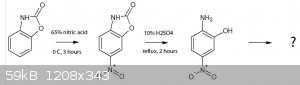
[Edited on 2-9-2015 by nitro-genes]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
| Quote: Originally posted by nitro-genes |
@ At Rosco, haha amazing, had the same idea and wanted to try it once! I never
got around to finding a proper synthesis protocol for dinitrosalicylic acid, so thanks for the references. Also wondered how much easier the dinitro
salicylic acid would reduce one nitro group, as opposed to trinitrophenol. Every TNP reduction scheme, other than with either commercial or homemade
hydrogensulfide, seems to result in an mess from which only small amounts of picramic acid can be isolated. But indeed, this may be worth a try, I
read the first reference you provided, and it indeed seems more selective than picric acid. There can be a big difference though between umol
quantities as quantitative test and a high yield synthesis. Same goes for ascorbic acid and picric acid, which seems to form some form of inseparable
complex with either picric acid or picramic, I read an article on this, but can't find it again, sorry.
[Edited on 2-9-2015 by nitro-genes] |
It is interesting if the route to o-DDNP from aspirin could be simplified to only require a dinitration condition, and would proceed at cooler
temperatures. I don't know for sure but I think the reduction using sugar is selective for the one nitro and is not prone to further reduce the
dinitrosalicylic acid to a diaminosalicylic acid. Most references only describe the color change as the point of interest for colorimetry tests of
sugar content determinations done by titration to color endpoint for glucose solutions, where there is no actual isolation done for the
3-amino-5-nitrosalicylic acid which is identified as responsible for that color change that is produced by the sugar reduction of the dinitrosalicylic
acid sodium salt.
Similarly there is a sugar determination by colorimetry possible using picric acid or actually it is sodium picrate being reduced in alkaline
condition by glucose to form picramic acid sodium salt. Likewise for those reduction methods, there is described no actual isolation of the picramic
acid. The reduction occurs in dilute solutions so the colorimetry methods may not be best for workup of more concentrated reaction mixtures where the
desired result is isolation of the crystallized product of the reduction. The solubilities of the materials involved will govern what are the most
desirable methods for reduction. Use of sulfides can be avoided even for the reduction of sodium picrate so those are candidate reduction schemes
which may be adaptable to the reduction of dinitrosalicylic acid sodium salt, if glucose proves not to be desirable for use when the product of
reduction is wished to be isolated as a solid, instead of only wished to produce a color change in a solution.
Many are the joys of dye chemistry! The fun just never ends!
Some of these dyes have such penetrating properties that they can stain a hole right through armor plate.
[Edited on 9/3/2015 by Rosco Bodine]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Viable route to a possible iso-DDNR? What do you guys think?
Up to the reduction of 5-nitro benzoxazolone to 5-amino benzoxazolone is described in literature, the nitration of the latter isn't, so this is
somewhat of a guess that the 4,6 nitro derivative would result, same for the oxidation/diazotization.
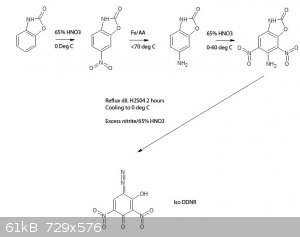
[Edited on 7-9-2015 by nitro-genes]
|
|
|
greenlight
National Hazard
Posts: 734
Registered: 3-11-2014
Member Is Offline
Mood: Energetic
|
|
@Nitro genes,
I am going to have a go at your DDNP synthesis from acetaminophen over the next week or so. I have already extracted the acetaminophen from homebrand
panadol.
For the second step, when nitrating to dinitro-acetaminophen, would 70% Nitric acid be more efficient. I have lots of Nitric but not much Ammonium
nitrate.
How many ml of 70% Nitric acid would you (or anyone) suggest to use in place of each gram of Ammonium nitrate?
Be good, otherwise be good at it
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
70% nitric acid means 70 grams of HNO3 per 100 grams, the molecular weight of HNO3 is 63, that means 1.11 moles of HNO3 per 100 grams of 70% nitric.
The molecular weight of acetaminophen is 151.2, since we need about a 2.2 molar equivalent to obtain the dinitroacetaminophen, we need (2.2/1.11)*100=
198 grams of 70% nitric to nitrate 1 mole or 151.2 grams of acetaminophen. Density of 70% nitric acid at 20C = 1.4134 --> so we would need
198/1.4134 = 140 ml of 70% nitric.
For 20 grams of acetaminophen, we need (20/151.2)*198= 26.2 grams 70% nitric, or 26.2/1.4134 = 18.53 ml
If we rougly use the figures from patent GB24409 for the sulfuric acid, the synthesis would look something like this:
- Pour 27 grams (15 ml) of 96% sulfuric acid in a small 100 ml beaker, and slowly add 26.2 grams (18.5 ml) of 70% nitric acid, while keeping
temperature below 20 deg C.
- In a seperate beaker on ice, dissolve 20 grams of finely powdered acetaminophen in 75 grams (41 ml) 96% sulfuric acid at <10 deg C.
- When all the acetaminopehn is dissolved, slowly add the sulfuric acid/nitric acid in 2 ml portions, while keeping temp around 5 deg C.
- After the last addition, keep stirring on ice for 1 or 2 hours.
Etc...
Good luck and keep us posted!
Regarding the synthesis of 3-azido 2,4,6 trinitrophenol as a precursor to the benzofuroxan posted earlier:
US 8748639 B1
3-azido-2,4,6-trinitrophenol, method of making, and method of transforming
12 mL of concentrated sulfuric acid were charged to the flask depicted in the experimental setup, and agitation was initiated. 0.897 grams (0.0037
mol) of 3-APA was then added and rinsed down with an additional 3.5 mL of concentrated sulfuric acid. The 3-APA dissolved readily and heating the
reaction mixture was unnecessary. The resultant solution was cooled to 3° C. using an ice water bath, and 0.39 grams (0.0057 mol) of NaNO2 dissolved
in 6 grams of concentrated sulfuric acid was added dropwise, keeping the temperature below 5° C. This addition took less than 5 minutes. The
resultant solution was then stirred for 20 minutes at that temperature. 25 mL of phosphoric acid was then added at such a rate that the temperature
did not exceed 5° C. This addition was somewhat exothermic and occurred over 45-50 minutes. After addition was complete, the reaction mixture was
stirred at less than 5° C. for 10 minutes. During the addition and subsequent stirring, the reaction mixture became quite viscous and attained a
pink-orange color. 1.18 grams (0.018 mol) of NaN3 was then added in portions, keeping the temperature below 5° C. This addition took less than 5
minutes. The reaction mixture was then stirred at less than 5° C. for 20 minutes, and was then allowed to come to room temperature by removing the
ice water bath. Once the reaction mixture had reached room temperature, it was poured into 70 mL of cold (<5° C.) water with stirring. The
resultant yellow precipitate was then collected by filtration, and washed with 2×20 mL of cold water. The yellow solid exhibited some water
solubility, so the water washed were retained for further analysis and/or crystal growth. The remaining precipitate was then dried in a dessicator,
and yielded 0.44 grams (44% yield)."
The following patent Patent US2129136 may be interesting, possibly the ethylsulfate diazonium salt of 3-diazo 2,4,6 trinitrophenol may be formed as a
reasonably stable intermediate, possibly circumventing the synthesis abovem which would be unsuitable for scaling up IMO.
Instead of adding phosphoric acid and NaN3 to the sulfuric acid/diazozium mixture, ethanol may be added slowly in order to form the ethylsulfate in
situ and precipitate the ethylsulfate diazonium salt, which may be reacted with 2 equivalents of hydrazine directly at low temperatures to produce the
benzofuroxan. Even better would be the reaction of calcium hydroxide or carbonate and hydrazine sulfate.
[Edited on 8-9-2015 by nitro-genes]
|
|
|
greenlight
National Hazard
Posts: 734
Registered: 3-11-2014
Member Is Offline
Mood: Energetic
|
|
Thanks for that nitro-genes, I will use those figures.
I will do the nitration soon and post results.
Be good, otherwise be good at it
|
|
|
greenlight
National Hazard
Posts: 734
Registered: 3-11-2014
Member Is Offline
Mood: Energetic
|
|
I just tried Nitro-genes dinitration of acetaminophen but used 70% Nitric acid instead of ammonium nitrate but something weird happened at the end.
I dissolved the Acetaminophen in 41 ml 98% Sulfuric acid and made the nitrating mix without going over 20 Degrees.
Cooled the Acetaminophen/Sulfuric acid to below 5 Degrees and started to add the nitrating mix in small portions. It responded in a temp rise very
quickly going up a degrees when 4 drops were added at the start and then after about 20 drops halfway through.
The mixture attained a dark brown colour when half the nitrating mix was added as was stated in the synth. The colour started to fade to orange after
the halfway point.
When there was only about 10-12 ml of nitrating mix left to add, yellow precipitate started to build up almost instantly and stopped the stir bar from
even being able to spin because it got so thick so fast as can be seen in second picture. After a minute the whole thing was just a mass of fine
crystal soup.
As it couldn't stir automatically anymore, I stirred by hand for another 5 minutes and then added the crushed ice which can be seen in the third and
fourth pictures.
It looks like what dinitroacetaminophen has been described to look like and dyes the skin yellow on contact.
Still useable even though the last bit of nitrating mix was not added?
[Edited on 11-9-2015 by greenlight]
   
[Edited on 11-9-2015 by greenlight]
Be good, otherwise be good at it
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Hi Greenlight,
The nitration is exceedingly fast, I think most of the dinitroacetaminophenol present suddenly all precipitated from the nitration mix in the form of
presumably small needle shaped crystals, making the mix very hard to stir. I've seen the same happening with AN, though not to this extent, which may
be a propertie of the added AN, or extra SA present. For the synthesis I posted in the prepublication section I also added the remark that with
minimal sulfuric acid present, the mix becomes very viscous and difficult to stir, maybe for the next time the acetaminophen is better dissolved in
slightly more sulfuric acid for the nitration using 70% nitric acid, say 100 grams instead of 75 grams. Alternatively, it may help to keep the
nitration mix at 10 degrees C after half of the SA/nitric acid is added. Anyway, I would just continue, the only uncertainty is how much of the
mono-nitro compound is present in the precipitate.
|
|
|
| Pages:
1
..
19
20
21
22
23
..
33 |
|








