Synthesis of ethyl acetylferulate
Recently I asked for help on a bromination reaction not going as expected (http://www.sciencemadness.org/talk/viewthread.php?tid=154654)
I have yet to get to the bottom of that issue, but it was pointed out by AvBaeyer that the method I used for acetylation was not very good. I needed
to produce more of the ester product, so looked for alternatives in the literature. I wanted to avoid buying acetic anhydride and pyridine, both of
which have availability issues for the home experimenter. I have described the process I used below.
This is the reaction sequence:
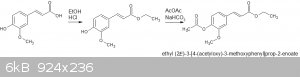
Many of the physical properties of the products, and methods for synthesis can be found in two reports from "The Institute of Paper Chemistry". The
paper titles are "Preparation of several lignin model compounds in the C6--C3 Series", and "Further work on the preparation of lignin model compounds
in the C6--C3 series", Irwin Pearl, 1956 and 1957.
The first step is preparation of the ethyl ester. I used a method as per "Acetyl chloride-methanol as a convenient reagent for...carboxylate ester
formation...", Nudelman et al, 1998.
26.71g of ferulic acid (0.138mol) was dissolved in 300ml of EtOH, resulting in a dark gold/brown solution.
I think my ferulic acid is not so pure. This is a different batch than I used previously, the solution should be a light yellow colour.
Recrystalisation of the ferulic acid from hot water is advised in future.
To an ice-cold solution of ferulic acid in EtOH, 0.18 mol / 14g of acetyl chloride was added drop-wise, forming EtOAc and HCl in solution. After the
last drop was added, the solution was allowed to stir at room temperature for 18 hours.
The EtOH and EtOAc was removed at 40C and reduced pressure (using my BRAND NEW ROTOVAC  ) and the remnant re-dissolved in another 100ml of EtOH and another 14g of acetyl chloride and reacted again for a further 18 hours, after
which the solvent was again removed, and washed/evaporated twice more to remove the last remnants of HCl. ) and the remnant re-dissolved in another 100ml of EtOH and another 14g of acetyl chloride and reacted again for a further 18 hours, after
which the solvent was again removed, and washed/evaporated twice more to remove the last remnants of HCl.
The result was a dirty dark yellow-green-brown oil. The literature indicates this should be a light yellow oil, so again, I suspect my source product.
The ester solidified spontaneously overnight into a yellow-green crystalline mass. I identified no solvents suitable for recrystalising it, so used it
in the next step as-is.
There are alternative esterification methods in the Pearl papers using sulfuric acid. This may turn out to be preferable, purely as I have noticed
already some corrosion on metal surfaces from the HCl.
For the acetylation step, I used the approach described in "Facile and efficient acetylation of primary alcohols and phenols with acetic anhydride
catalyzed by dried sodium bicarbonate", Catalysts, Lugemwu, Shaikh, Hochstedt, 2013.
The raw ethyl ester was dissolved in 150ml of toluene resulting in a yellow-gree solution with some insoluble matter noted. This solution was filtered
before use.
Separately, acetic anydride was prepared in-situ. 38.8g acetyl chloride (0.49mol) was added dropwise to a cold suspension of 41g (0.5 mol) of sodium
acetate in 80ml of toluene, and stirred at room temperature for 30 minutes. The ester solution was then added all at once, and 16.8g NaHCO3 (0.2mol)
was added. A very small amount of gas was evolved on addition.
This mixture was stirred for 24 hours at room temperature. TLC (1:3 EtOAc:hexane) after 24 hours showed a single spot at Rf = 0.51. In comparison, the
ester Rf is 0.43, and the acid barely moves off the start line.
The mixture was filtered. The solids retained quite a large amount of solvent, so was manually pressed to squeeze out more solvent. The final solid
remaining were washed into water, resulting in a cloudy aqueous layer and a scummy looking yellow solution floating on top. This was left for 24 hours
and the toluene evaporated, leaving suspended off-white product. This was dried then recrystalized from EtOH.
The filtrate of toluene solution of the bulk of the product was removed at 60C under reduced pressure, with the product eventually solidifying in the
flask. EtOH was used to dissolve the product to remove it from the flask, and for recrystalisation.
After cooling to room temperature, and then refrigerated, the main bulk of the product from the toluene solution crystalized out as tiny white
needles. Yield was 22.82g, with a further 1.95g recovered from the solids.
Melting point was measured at 120 - 121C, lit. 122-123C.
Total yield of 24.8g, at M = 264.27, thus about 94mmol, or a yield of 68% on the original source ferulic acid.
So yes, a greatly improved yield using acetic anhydride. Thanks, AvBaeyer.
I will use this material to pursue the bromination step again, and the Pearl papers have also given me ideas towards reduction of the double bond,
avoiding the use of Palladium/high pressure/hydrogen etc.
|









