| Pages:
1
2 |
Nicodem
Super Moderator

Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
Trimethylsulfonium bromide and its use for the methylation of phenols
Recently, there was some discussion on the forum about using trimethylsulfonium bromide (Me3SBr) as an electrophilic methylating reagent [1]. So I did
a literature search on the use of sulfonium salts as alkylating reagents for various types of nucleophiles [2]. There are quite numerous examples of
their use in organic synthesis, though what I found was mostly focused on the more reactive dialkylarylsulfonium or alkyldiarylsulfonium salts and
asymmetric C-alkylation of enolates (with chiral sulfonium salts). There were described some methylations with trimethylsulfonium hydroxide (Me3SOH),
but I could find nothing with Me3SBr. I decided to prepare this reagent and test its methylating ability.
Me3SBr was prepared by the reaction of bromine with dimethylsulfoxide (DMSO) by a simplification of a published method [3]. Other methods from DMSO
are also known (e.g., by the reaction with benzyl bromide or ethyl bromoacetate [1]). Reaction of DMSO with hydrogen bromide might also lead to
Me3SBr, but I could find no literature support for this. Trimethylsulfonium chloride (Me3SCl) analogously forms from the reaction of DMSO with Cl2,[4]
but I could find no specific information on the selectivity and no preparative example.
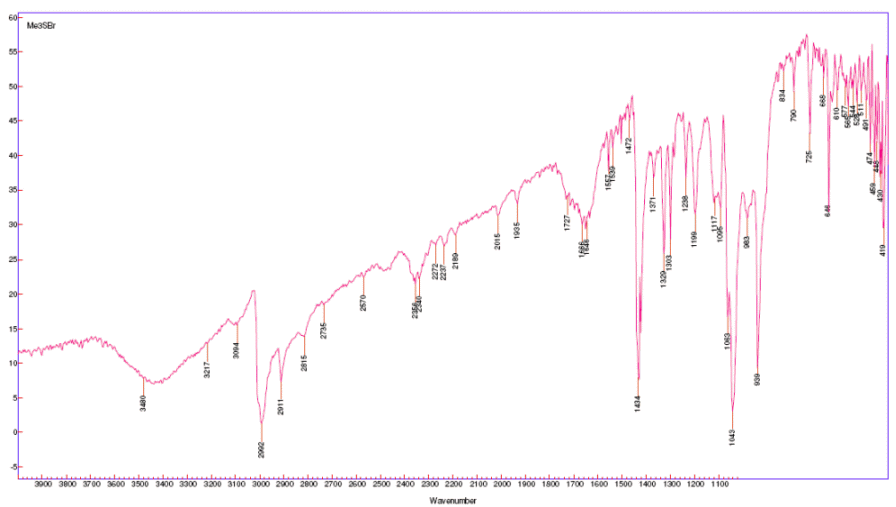
Trimethylsulfonium bromide (Me3SBr):
Into a 20 mL dimethylsulfoxide (DMSO) was slowly and very carefully added 2 mL bromine (39 mmol) dropwise from a micropipette, while stirring in a
cooling bath (note 1), at such a rate to maintain the temperature of the reaction mixture at about 40 °C. The reaction is very exothermic and each
drop of Br2 makes a hissing noise upon contact with DMSO. The clear yellow solution was left stirring for 3 days at room temperature. It formed a
thick yellow slurry which was then diluted with 60 mL acetone. The insoluble product was vacuum filtered, washed with 3×15 mL acetone and left drying
under vacuum in a desiccator. There was thus obtained 7.72 g (63%) of a light yellow crystalline powder having a faint “sulfide stench” (note 2):
mp 194–198 °C, subl. (lit.[3] 198–200 °C, from EtOH); IR (KBr):
Note 1: The oxidation of DMSO with Br2 is extremely exothermic! Each drop of Br2 would violently react with
DMSO (similarly as adding TCCA to DMSO). Adding all the bromine at once can result in accidents and serious injuries! Cooling and good stirring is
advised. Do not use an ice bath (mp of DMSO!), but a water bath at about 15 °C will do. Do not upscale the reaction without all the necessary
precautions!
Note 2: During the reaction there were no gasses evolved. During the work up some dimethyl sulfide smell was present but not up to any annoying level.
Use of Me3SBr
I looked on the shelves for a phenolic substrate to O-methylate which would be less reactive (e.g., having a hydroxy orto to a carbonyl
group) and giving a product that would be solid at room temperature (so that no chromatography would be needed in the reaction work up). I found some
old 2-hydroxy-5-methylbenzophenone from Aldrich picking dust on the shelves, which looked like nobody will ever use or miss. I decided to sacrifice it
in the name of amateur chemistry.
Not being sure about the reaction temperature required, or best solvent to use, I decided not to waste time reinventing the wheel and used the
conditions previously reported to work for tetramethylammonium chloride as a reagent for the methylation of phenols [6]. Since sulfonium salts are
more electrophilic than the ammonium counterparts, I used a lower reaction temperature (100 °C instead of 150 °C). It worked just fine.
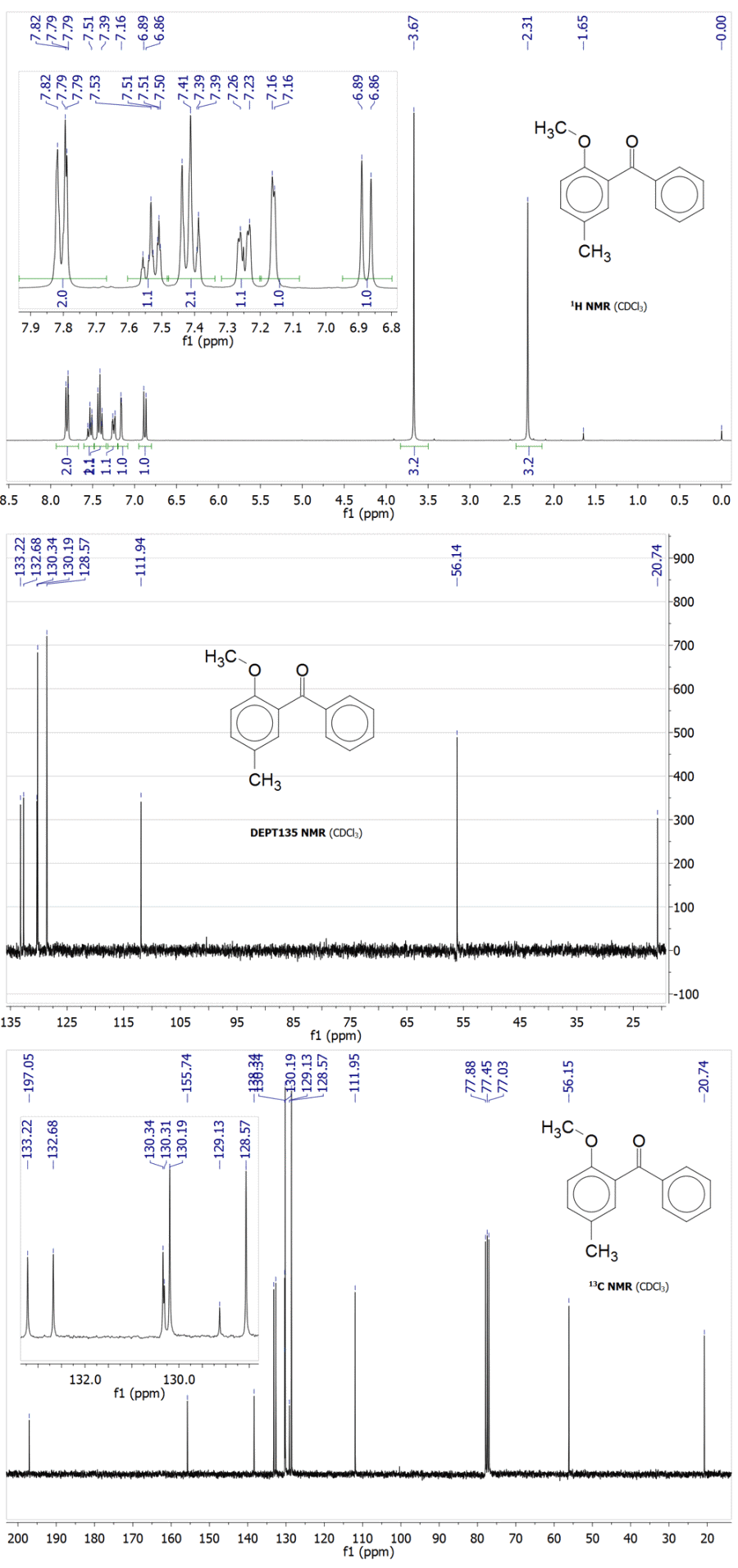
Methylation of 2-hydroxy-5-methylbenzophenone with trimethylsulfonium bromide:
A mixture of 1.90 g Me3SBr (12 mmol), 2.12 g 2-hydroxy-5-methylbenzophenone (10 mmol), 1.66 g K2CO3 (12 mmol) and 6 mL polyethyleneglycol (PEG400) was
stirred for 6 h at 100 °C (note 1). The reaction was checked with TLC at 3 h and there was only one spot for a new product being formed, while the
spot of the starting phenol was already faint. The reaction mixture was then worked up by diluting in 100 mL water and extraction into 40 mL
diisopropyl ether, which was then washed with 3×40 mL 1M NaOH(aq), 100 mL water and rotavaped to give 2.08 g (92%) of a TLC pure colorless viscous
oil. This was crystallized by dissolving it in 30 mL methanol, precipitating the oily product with 10 mL water and leaving this mixture overnight at
-16 °C. The solidified product was vacuum filtered and dried under vacuum in a desiccators to give 1.93 g (85%) of 2-methoxy-5-methylbenzophenone as
chunks of solids with part of it having crystallized nicely in needles: mp 37–38 °C (lit.[5] 37–38 °C); NMR (300 MHz, CDCl3):
Note 1: During the course of the reaction there was dimethyl sulfide forming as perceived by its smell. But since this was done in a fume hood this
represented no problem. On such a small reaction scale and such a slow reaction rate, this is no problem even out of the fume hood as long as your
neighbors don’t mind, but on a larger scale I suggest to recover the dimethyl sulfide by absorption in acetone.
References
[1] https://www.sciencemadness.org/whisper/viewthread.php?tid=15...
[2] (a) K. Yamauchi, T. Tanabe, M. Kinoshita, J. Org. Chem., 44, 1979, 638–639, DOI: 10.1021/jo01318a037 (b) M. Kobayashi, K.Umemura, N.
Watanabe, H. Matsuyama, Chemistry Letters, 1985, 1067–1070. (c) M. Kobayashi, K.Umemura, H. Matsuyama, Chemistry Letters, 1987,
327–328. (d) K. Umemura, H. Matsuyama, N. Watanabe, M. Kobayashi, N. Kamigata, J. Org. Chem., 54, 1989, 2374–2383, DOI:
10.1021/jo00271a025 (e) K. Umemura, H. Matsuyama, N. Kamigata, Bull. Chem. Soc. Jpn. 63, 1990, 2593–2600 (f) N. Shibata, A. Matsnev, D.
Cahard, Beilstein J. Org. Chem., 6, 2010, 65, DOI: 10.3762/bjoc.6.65
(these are not a result of a full literature review, but see also the references therein)
[3] D. Martin, A. Berger, Journal für Praktische Chemie, 312, 1970, 683–689, DOI: 10.1002/prac.19703120418
[4] J. R. Gauvreau, S. Poignant, G. J. Martin Tetrahedron Letters, 21, 1980, 1319–1322.
[5] G. Stadnikoff, A. Baryschewa, Ber., 61, 1928, 1996–1999, DOI: 10.1002/cber.19280610869
[6] http://www.sciencemadness.org/talk/viewthread.php?tid=3230&a...
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
smuv
National Hazard
Posts: 842
Registered: 2-5-2007
Member Is Offline
Mood: Jingoistic
|
|
Good, I am glad you tried this. It would be nice to see what this can do in more mild conditions, say refluxing acetone.
It's a good yield too. I have had slow reactions alkylating ortho acyl phenols in the past, even with an alkyl bromide.
Oh and about the proton, looks more like an NMR of CDCl3 in your ether than the other way around  But I do that too for non critical products when I don't care about seeing coupling nuances and also need to take a
carbon. But I do that too for non critical products when I don't care about seeing coupling nuances and also need to take a
carbon.
BTW was the starting material yellow?
|
|
|
bbartlog
International Hazard
Posts: 1139
Registered: 27-8-2009
Location: Unmoored in time
Member Is Offline
Mood: No Mood
|
|
Nice. Seems like a fairly easy reagent to prepare, too. What is the reason for the three days' stirring following the addition of the bromine to the
DMSO? I assume there is some initial (fast, exothermic) reaction followed by a second slower one?
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
I'm not sure any more that the bp of acetone would allow for an acceptable rate of the reaction. I might try (time allowing) some higher boiling
solvent (something aprotic and not too polar with bp 70-80 °C), but I think I will have to use a more reactive phenol than the above tested one.
| Quote: | | It's a good yield too. I have had slow reactions alkylating ortho acyl phenols in the past, even with an alkyl bromide. |
Yes, phenols having an ortho carbonyl substitution (acyl, formyl, ester, etc.) are notoriously slow to alkylate. For example, you can find
examples in the literature, where O-alkylation of 2,4-dihydroxyacetophenone and related substrates is selectively performed at the 4-hydroxy group
which indicates the 2-hydroxy is at least one or two magnitudes less reactive.
| Quote: | | Oh and about the proton, looks more like an NMR of CDCl3 in your ether than the other way around But I do that too for non critical products when I don't care about seeing coupling nuances and also need to take a
carbon. |
That's what you do when you are in hurry to take the spectra. I did not weight the sample, but I would judge I used about 50-100 mg in 0.7 mL CDCl3.
This is why the 13C NMR took no more than 100 scans, but I left it go longer for cosmetic reasons. In any case, the concentration of the sample does
not have much of a negative influence on 1H spectra - unless you have exchangeable protons, shifts usually only vary slightly and couplings don't
alter.
| Quote: | | BTW was the starting material yellow? |
Yes, the 2-hydroxy-5-methylbenzophenone is a canary yellow crystalline compound (tinny needles). The methylated product is white (colourless
actually). Seems like the O-methylation breaks some chromophore system (The H-bonding of OH with CO keeping the whole system in conjugation?).
Quote: Originally posted by bbartlog  | | Nice. Seems like a fairly easy reagent to prepare, too. What is the reason for the three days' stirring following the addition of the bromine to the
DMSO? I assume there is some initial (fast, exothermic) reaction followed by a second slower one? |
Yes, Me3SBr is ridiculously easy to prepare, but this is not a safe procedure for the beginners who always tend to ignore the safety and do reckless
things. Bromine is not a suitable reagent for safe use in such hands. It worries me that someone tries to repeat this without taking proper
precautions. Mixing oxidants and reducers is not for the negligent.
Anyway, yes the S-bromination of DMSO is probably instantaneous (hence the violent reaction) and the Pummerer rearrangement is also fast at such
temperatures. It is probably the following disproportionation of DMSO to give Me2S, among other things, and finally Me3SBr, that is the slow part of
the reaction (otherwise, the proposed mechanism of the overall reaction is nicely described in the reference [3]).
One or two days would probably do fine, but the "3 days" was the weekend factor. 
The procedure in [3] was not really aimed to be preparative. It is performed in a way to recover all the reaction products for analysis (not only
Me3SBr which is what I was after). It also called for 5 h heating at 50 °C, while I prefer reactions at room temperature that one can just set,
forget for a while, and then work up when time allows. Hence I simplified it in a way to suit my needs.
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
smuv
National Hazard
Posts: 842
Registered: 2-5-2007
Member Is Offline
Mood: Jingoistic
|
|
I was just giving you a hard time nicodem, everyone does it on occasion. In addition to resolution of exchangable protons, you can hide impurities by
taking protons of such concentrated samples because the peaks become very broadened; reviewers can give you a hard time because people use it as a sly
trick to make a so-so product look like a very pure one.
Ya, about the color, thats probably it, deprotonation of the phenol to cause it to exist in some extent as a ketone (and enolization of the acyl
ketone). I read somewhere that in solution and/or solid state (can't remember exactly) these systems exist to a noticable extent as the intraolecular
hemiacetal. Both 2-hydroxy-5-methoxyphenacyl chloride and 2-hydroxy-5-methoxyacetophenone are yellow and their corresponding 2-ethoxy derivatives are
colorless. Looks like the same is true in this case as well.
Anyways, it is nice to see reaction development on sciencemadness.
[Edited on 3-5-2011 by smuv]
|
|
|
madscientist
National Hazard
Posts: 962
Registered: 19-5-2002
Location: American Midwest
Member Is Offline
Mood: pyrophoric
|
|
Great work Nicodem! It's nice to know we can methylate phenols in an amateur environment without dealing with dangerous carcinogens.
I weep at the sight of flaming acetic anhydride.
|
|
|
spirocycle
Hazard to Others
Posts: 197
Registered: 29-9-2010
Member Is Offline
Mood: No Mood
|
|
^are you suggesting that trimethyl sulfonium bromide is NOT carcinogenic?
is this a fact or an assumption?
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
But why would you suspect trimethylsulfonium salts could be carcinogenic?
With such preemptive assumptions you could just as well label any chemical as potentially carcinogenic, which would be result in completely insane
behaviour (like any preemtive actions usually do).
Me3SBr is too small and unreactive to be considered active in any of known mechanisms of genotoxicity. A TOXNET search on "trimethylsulfonium" gives
nothing indicative of any such dangers. The MSDS of Me3SBr is unfortunately void of toxicological data which is indicative that there never were any
such studies, however the MSDS of trimethylsulfonium iodide says in regard to carcinogenicity: "No component of this product present at levels greater
than or equal to 0.1% is identified as probable, possible or confirmed human carcinogen by IARC." This does not mean that trimethylsulfonium salts
were ever confirmed to genotox free, but it at least says that nobody considers them as potentially genotoxic, if nothing else at least because they
do not fit any theoretic criteria.
I would be more worried about the usual acute toxicity. Such onium salts are usually ion channel blockers and thus can be quite toxic by ingestion.
(not that I expect anybody is going to ingest them)
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
spirocycle
Hazard to Others
Posts: 197
Registered: 29-9-2010
Member Is Offline
Mood: No Mood
|
|
is it really that odd to assume a methylating agent could be carcinogenic?
arent many (if not most) methylating agents carcinogenic?
MeI is smaller than Me3SBr, so that isnt really a valid point.
if it is so unreactive, then why is it a valid alternative to other methylating agents?
|
|
|
smuv
National Hazard
Posts: 842
Registered: 2-5-2007
Member Is Offline
Mood: Jingoistic
|
|
Thats a narrow minded way of looking at things, there are so many aspects to carcinogenisis that blanket statements like all methylating agents are
carcinogens will always be false.
Methyl bromide for example, if even a carcinogen is a very mild one (it has not been consistently proven to be so in even animal studies).
[Edited on 3-19-2011 by smuv]
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
With "too small", I had in mind the genotoxicity mechanisms relying in direct non-covalent interaction with the chromosomes, disruption of mitotic
processes, formation of reactive metabolites binding to the DNA (aflatoxines, benzene, etc.), or any such similar, because these activities are
unlikely to reside in such simple molecules. Small genotoxic molecules are practically exclusively electrophilic alkylating reagents or otherwise able
to react with amino groups of the nucleic bases, but this is not really applicable to trimethylsulfonium salts due to its low electrophilicity (if it
was, then S-methylmethionine would be dangerous as well).
| Quote: | | is it really that odd to assume a methylating agent could be carcinogenic? |
The methylating ability is no evidence of genotoxicity, and even less so of carcinogenicity (not every genotoxic compound is carcinogenic, and not
every carcinogenic compound is a known human carcinogen). For example, MeBr and MeI have confirmed mutagenic activity, but MeCl was found inactive in
the same test. This however does not necessarily means that MeI and MeBr are also carcinogenic, but since they were found to posses at least one
genotoxic activity, they should be considered as potentially carcinogenic. Though, you will probably never live to hear about MeI being a known
human carcinogen for the simple reason, that to gain this title, there must exist statistical evidence. However, since MeI is not used
industrially (way too expensive!), at least not in large amounts and continuously enough for a statistical evaluation, there can not be any such study
upgrading it to this top level title. Yet, its industrially useful alternative, dimethyl sulfate, got to the IARC 2A category without troubles.
So there is this requirement for statistical evidence, which means that a compound can only be labelled as a known human carcinogen if it is, or was
used, in the industry for a large enough period of time. This can cause some extremely paradoxical situations. For example, methoxymethyl chloride is
a known human carcinogen (lots of workers in the chemical industry died from it) and is now prohibited from transportation and nearly impossible to
buy even for the academic research. Due to its reclassification, methoxymethyl bromide is ofered as an alternative reagent for the introduction of the
MOM protecting group and other uses. But we know from the MeCl < MeBr < MeI ~ (MeO)2SO2 ~ MeOMs < MeOTf series that the more reactive the
methylating reagent the more genotoxic it is, so what are we suppose to think about the lack of warnings regarding methoxymethyl bromide handling?
What about the students and researchers using it without any proper care and protection? I guess we have to wait for a statistically relevant sample
to confirm the associated morbidity.
But don't wait for change of classification to be prudent. If a compound is genotoxic, you should consider it mutagenic and potentially carcinogenic -
so treat with all due respect and avoid exposure, just to be on the sure side.
| Quote: | | if it is so unreactive, then why is it a valid alternative to other methylating agents? |
Ironically, the answer is already in your question. It is a valid alternative to other methylating reagents exactly because it is so unreactive. The
situation here is similar as for the industry where you have the non-genotoxic unreactive dimethyl carbonate vs. genotoxic reactive dimethyl sulfate.
Except that we are dealing with the amateur issues, so that we get to discuss the non-genotoxic unreactive onium salts vs. genotoxic reactive dimethyl
sulfate and methyl iodide.
[Edited on 19/3/2011 by Nicodem]
|
|
|
spirocycle
Hazard to Others
Posts: 197
Registered: 29-9-2010
Member Is Offline
Mood: No Mood
|
|
| Quote: | | But we know from the MeCl < MeBr < MeI ~ (MeO)2SO2 ~ MeOMs < MeOTf series that the more reactive the methylating reagent the more genotoxic
it is |
| Quote: | | It is a valid alternative to other methylating reagents exactly because it is so unreactive. |
Thanks for the info, but I still have one more question based on the above quotes:
so you are saying that TMSB is less reactive, making it less harmful? Would this imply that the reaction times would be significantly greather than
with a more reactive methylating agent?
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
Only in the sense that it is unlikely to be genotoxic and thus highly unlikely to be carcinogenic.
| Quote: | | Would this imply that the reaction times would be significantly greather than with a more reactive methylating agent? |
Yes, of course. Just check the reaction conditions I used. Compare that to the reactivity of dimethyl sulfate or methyl iodide, both of which
efficiently methylate most phenols at room temperature when using the same base in acetone or acetonitrile as solvents. This makes Me3SBr a few
hundreds times less reactive, but as you can see this is no obstacle for its use even on the less reactive phenols (100 °C is still acceptable for
most phenolic substrates).
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
spirocycle
Hazard to Others
Posts: 197
Registered: 29-9-2010
Member Is Offline
Mood: No Mood
|
|
thanks for clearing that up
|
|
|
spirocycle
Hazard to Others
Posts: 197
Registered: 29-9-2010
Member Is Offline
Mood: No Mood
|
|
Just a note for those trying this:
when diluted with acetone, the mix becomes a potent lachrymator
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
I noticed no such thing, though I certainly did not put my nose into it.
For how long did you let the reaction run? What was the reaction temperature profile? What colour was it when you quenched it? What was the yield?
|
|
|
organometallic
Hazard to Self
Posts: 53
Registered: 22-7-2007
Location: United Kingdom
Member Is Offline
Mood: No Mood
|
|
Hi Nicodem,
A while back you said you'd also tested this methylating agent on vanillin .. Please do give us a few words re yeild and observations about that if
you have the time.
I intend to try this on vanillin and also 5-hydroxyvanillin
[Edited on 12-5-2011 by organometallic]
In vials of ivory and coloured glass
Unstoppered, lurked her strange synthetic perfumes,
Unguent, powdered, or liquid - troubled, confused
And drowned the sense in odours.
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by organometallic | Hi Nicodem,
A while back you said you'd also tested this methylating agent on vanillin .. Please do give us a few words re yeild and observations about that if
you have the time.
I intend to try this on vanillin and also 5-hydroxyvanillin |
I certainly will not be doing any further experiments on this topic in the near future, as I'm running out of time even on things I must do. I did
once set up the reaction for methylation of vanillin, but never got the time to analyse or work up, so it went in the waste. But I do encourage you to
do some experiments. Let us know how it went. Not only about the methylations, but also on the synthesis of Me3SBr. I'm especially interested to hear
about any (safely performed) scale-up experiments.
In the methylation step, you might also try some solvents other than PEG. PEG is cheap, widely available and probably optimal, but it would be still
interesting if it turns out that some substrates could be methylated neat, or in higher boiling alcohols like 1- or 2-butanol, or in DMF, NMP, DMSO,
propylene glycol, some cellosolves, or related&available solvents. There is plenty room for experimentation.
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
aliced25
Hazard to Others
Posts: 262
Registered: 31-7-2010
Member Is Offline
Mood: No Mood
|
|
Just a quick question, in relation to a hypothetical matter - what chance would you give the trimethylsulfonium bromide of effectively O-methylating
the p-hydroxyl in N-acetyltyrosine or N-protected tyramine?
From a Knight of the Realm: "Animated movies are not just for kids, they're also for adults who do a lot of drugs." Sir Paul McCartney
|
|
|
smaerd
International Hazard
Posts: 1262
Registered: 23-1-2010
Member Is Offline
Mood: hmm...
|
|
Excellent work nicodem!
Was the DMSO purified or dried prior to use? Most commercial preparations of the chemical have moisture and that classic rotting garbage scent.
Any idea on whether or not this compound could be stored simply or might it have a limited shelf-life?
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
Sorry for these late replies - didn't notice the questions earlier.
| Quote: Originally posted by aliced25 | | Just a quick question, in relation to a hypothetical matter - what chance would you give the trimethylsulfonium bromide of effectively O-methylating
the p-hydroxyl in N-acetyltyrosine or N-protected tyramine? |
With N-acetyltyrosine, the carboxy group could also be methylated. I can't really estimate the selectivity. With N-acetyltyramine, I would expect the
above method would likely work fine.
| Quote: Originally posted by smaerd | Was the DMSO purified or dried prior to use? Most commercial preparations of the chemical have moisture and that classic rotting garbage scent.
Any idea on whether or not this compound could be stored simply or might it have a limited shelf-life? |
The DMSO was used straight from the reagent bottle. I don't really remember how old it was, but I doubt it contained much dimethyl sulfide and thus
that scent typical for DMSO made in the older days when the production process was not yet optimal. Dimethyl sulfide is a reaction intermediate in
this synthesis of Me3SBr anyway. I would expect some moisture in DMSO should be tolerated.
I recently noticed a post at Hyperlab where this synthesis of Me3SBr has been optimized to repeatedly give 80-85% yields (see post #567957
by ophite in the thread Russian Hyperlab|Метилирование
фенолов...). According to the author, the key - among other things - is in maintaining a low
temperature during the long reaction (the reaction rate is consequently slower and the reaction can take a week or more - depending on the
temperature). Apparently the reaction can be monitored by simple color change observation.
Considering that both atoms in Br2 are used in the reaction and that this reaction proceeds trough a complicated sequence, the achieved
yield is quite extraordinary. Ophite made quite a number of these Me3SBr syntheses and reading his report is must for anyone
attempting this (especially his caution warnings should be taken seriously).
Additionally, he tried several interesting experiments using DMSO as the solvent for the methylation of hydroquinone with Me3SBr and with
NaOH as the base (see the Hyperlab post #567958 in that same thread). The methylations were successful, but gave poor to moderate yields of the
dimethylated product (28-46%). Most of the starting material got converted to the p-methoxyphenol.
DMSO would not be my first solvent choice for this reaction (for several reasons), so I'm surprised he even obtained that kind of yields. By
generalizing the conclusions about the methylation with the related onium salt Me4NCl described the DOI: 10.1016/j.tet.2008.10.024, the very polar solvents are a bad choice.
Ophite's experiment applying the same conditions to 5-methoxysalicylaldehyde gave no isolable product. As a negative result, this would be of
great interest in understanding the reaction, if a control experiment would be done applying the above described method (PEG and
K2CO3).
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
gauderbdf
Harmless
Posts: 2
Registered: 10-1-2013
Member Is Offline
Mood: No Mood
|
|
Hi everyone.
Some weeks ago I run the synthesis of Me3SBr following Nicodem's procedure with a modification which avoid handling of pure liquid bromine.
Unfortunately I didn't note the data of the experiment because this was just a trial.
However this is my procedure for the same Nicodem's scale.
Bromine was generated by slow addition of H2O2 to a water solution of KBr and H2SO4 at 0 °C, then it was extracted washing the mixture several times
(8-10) with n-heptane. All the extracts were mix together and dried over anhydrous MgSO4 overnight. The so obtained Br2 solution (Vtot around 400 ml)
was added to DMSO with no significant heating evolution and quick decoloration. Aftre 3 days (week-end) lower DMSO layer was recover and acetone was
addet to precipitate Me3S Br. Crystallization took place in some days yelding 1.91 g.
Anyone have a reference/known about the exact reaction and the reaction mechanism after bromination of DMSO (to calculate yield) ? I tried to
understend but I didn't.
P.S. I have a photo of productbut I don't understand how to attach it..help!!, thanks
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
Thanks for sharing the info.
| Quote: Originally posted by gauderbdf | | Bromine was generated by slow addition of H2O2 to a water solution of KBr and H2SO4 at 0 °C, then it was extracted washing the mixture several times
(8-10) with n-heptane. |
I think heptane does not efficiently extract bromine from the aq. solution. Using dichloromethane is a better option. Two extractions take out most of
the bromine. You also need to make sure there is a slight excess of hydrogen peroxide, as any excess of KBr will sequester bromine in the form of the
water partitioning tribromide anion.
However, dichloromethane and DMSO are miscible and the dillution could affect the reaction kinetics, its outcome, and Me3SBr solubility (it
might precipitate during the reaction, or the mixture would need to be concentrated before precipitating with acetone).
| Quote: | | Crystallization took place in some days yelding 1.91 g. |
On what amount of KBr?
| Quote: | | Anyone have a reference/known about the exact reaction and the reaction mechanism after bromination of DMSO (to calculate yield) ? I tried to
understend but I didn't. |
The proposed reaction sequence is in the article cited in the first post (the reference no. 3). Essentially, the stoichiometry is Br2 ->
2 Me3SBr, so that both bromine atoms participate.
| Quote: | | P.S. I have a photo of productbut I don't understand how to attach it..help!!, thanks |
You have an "Attachment:" field in the posting form where you browse for the file to attach. Just make sure you don't use the "Preview post" function,
else it resets the field. Though, I do not see any particular use of such a photo. A melting point analysis would be way more informative.
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
Mailinmypocket
International Hazard
Posts: 1352
Registered: 12-5-2011
Member Is Offline
Mood: No Mood
|
|
I am currently following Nicodem's procedure above, does anyone have a reference for the methylation of vanillin using trimethylsulfonium bromide? It
would be appreciated! And yes, the addition of bromine to DMSO is very exothermic, use caution if repeated.
*edit* the yield after filtering and washing with acetone is 5.98g. I'm assuming the lower yield is because I only let the reaction run for 25 hrs at
a somewhat low room temperature ~19c. The smell of rotten cabbage was becoming a bit noticeable despite the reaction flask being stoppered, not
extremely tightly mind you. I can confirm the Lacrimatory effects when acetone is added though, my eyes were burning like crazy, presumeably due to
bromoacetone formation and no time hood.
[file]29796[/]
 
[Edited on 16-2-2014 by Mailinmypocket]
|
|
|
dioxine
Harmless
Posts: 15
Registered: 17-12-2018
Location: Russia
Member Is Offline
|
|
It is okay to work with Me3SBr without strong fume hood? For sure i can tolerate the smell of Me2S, but there is little to no data about acute
toxicity of this compound. Is it posses any serious risks to eyes, respiratory system or something like that(I mean there are compounds that shouldn't
be even opened without fume hood, like Tetramethyl orthosilicate(TMOS) or DMS) or it's okay to handle it as something like bromine, hydrochloric
acid(easy to smell in small quantities, can be opened for a short time)?
Sorry for bad english.
|
|
|
| Pages:
1
2 |
|








