Diachrynic
Hazard to Others

Posts: 222
Registered: 23-9-2017
Location: western spiral arm of the galaxy
Member Is Offline
Mood: zenosyne
|
|
Dicyandiamide from calcium cyanamide fertilizer
Dicyandiamide is a useful precursor to many substances, particularly guanidine compounds, and can be easily and cheaply prepared from commonly
available calcium cyanamide fertilizer.
Chemicals:
Calcium cyanamide fertilizer (about 50% CaCN2, 15-18.3% N in form of calcium cyanamide)
Water
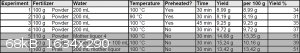
Multiple procedures have been tried for this and the yield doesn't seem to strongly depend on the exact times and temperatures, slight variation in
those affect the yield by about 4% up or down. The biggest yield improvement was found in reusing the mother liquor from one extraction for the next
one, increasing the yield from around 35% to 51%. The fertilizer comes in prilled form and it was used ground and unground with no effect on the
yield, so unground is preferred. The following is the best procedure out of the ones tried, but the alternative experimental conditions are summarized
in the table below:
Procedure:
100 g of fertilizer prills and 200 mL of deionized water are put into a 600 mL beaker and heated with stirring until boiling starts, this takes around
10-15 minutes depending on the hotplate. Once the mixture starts boiling it is kept there for 30 minutes while being covered with a watch glass. The
reaction is slightly exothermic and traces of ammonia are evolved which are barely detectable by smell but turn pH paper green-blue. The reaction
foams just slightly and using an adequately sized beaker and moderating the temperature once boiling starts keeps it under control. The reaction
doesn't have to agressively boil, but it should be around the boiling point. After the 30 minutes have elapsed the slurry is vacuum filtered while hot
over a büchner funnel, the mixture is easily filterable and doesn't clog the filter paper. The filter cake was washed with deionized water, total 60
mL in three portions, rinsing the mud out of the beaker into the filter. The slightly yellow filtrate is left to crystallize and finally cooled to 10
°C (which was the outside temperature at the time, but cooling to 0 °C would likely give a higher yield). Dicyandiamide crystallizes from this
solution in long, colorless needles, it is filtered off about 7 hours after the extraction. The filtrate usually turns brownish or greenish and is
kept for another extraction.
Yield: 9,72 g (37% assuming 50% calcium cyanamide content in the fertilizer)
When the mother liquor was topped up to 200 mL and used for another extraction, the yield was substancially higher. The increase in yield matches well
the calculated loss due to solubility in the first experiment. The mother liquors have been reused at least twice with no effect on the yield or
purity.
Yield: 13,42 g (51% assuming 50% calcium cyanamide content in the fertilizer)
Purification, melting point, test for identity:
The crude product was dissolved in distilled water and treated with ammonium oxalate solution, no turbidity was observed. This indicates that the
crude product contains little to no calcium.
A melting point test was conducted in a micro test tube in an oil bath. The melting point was 211-212 °C. This matches the literature value (it
should be noted that values from 205 °C to 214 °C have been reported, but 209-211 °C seem to be the most recent).
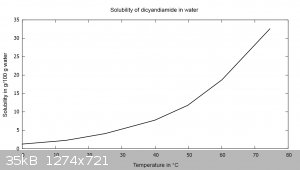
Dicyandiamide has a steep solubility curve in water and can be readily recrystallized from it. 81.58 g of dicyandiamide are dissolved in 400 mL of 75
°C water, filtered hot and cooled to 10 °C. The solution has a pH of 7-8. The crystal shape changes, it is no longer needles but rather rhombic
plate-like. Recovery was 70.06 g (86%). The previous tests show this is probably unnecessary. The reason for the change in crystal shape has not been
found so far. At 0 °C the solubility is 1.27 g/100 g water, at 74.5 °C it is 32.58 g/100 g water.
A test reported in the literature for dicyandiamide is boiling a sample with dilute sulfuric acid for a few hours, then adding copper sulfate and
dropwise adding ammonia or sodium hydroxide. This precipitates a rose-red (although I find it rather pastel red) copper complex of copper(II) and
guanylurea. When using sodium hydroxide the solid seems to be more brown-grey than with ammonia, but all the copper seems to be precipitated. It is
likely the brown color is due to lower complexes and hydroxospecies.
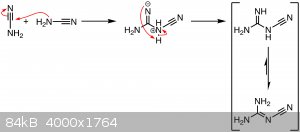
Mechanism:
Upon mixing the fertilizer in water and heating it, cyanamide is liberated from its salt and dimerizes to dicyandiamide due to the high pH (since the
fertilizer also contains lime) of the solution and the heat. Prolonged heating with strong, hot alkali decomposes dicyandiamide to many products under
evolution of ammonia, but this seems to negligeable in the conditions of my experiments.
CaCN2 + 2 H2O → Ca(OH)2 ↓ + H2NCN
Notes and cost:
The beakers used for the experiment have calcium deposits after the synthesis, these can be easily cleaned with dilute acetic acid.
Cost wise the process is very cheap. The fertilizer I used costed me 2.60 euros per kilo, which amounts to about 2-3 euros per 100 g of dicyandiamide.
This is on par if not slightly lower than commercial suppliers if you buy kilogramm amounts, at least the ones I have checked. From 1 kilo of
fertilizer about 130 g of dicyandiamide could be extracted.
Dicyandiamide can be used to make guanidine nitrate via melting with ammonium nitrate. This process is reported to give up to 95% yields, but I
haven't confirmed this as of now. Dicyandiamide can also be reacted with sodium azide and acid to produce 5-aminotetrazole in up to 97% yields, this
process is called the Schtolle method. For details see Engagers publication on tetrazoles and the original literature.
As a word of warning, calcium cyanamide fertilizer can be toxic to shallow rooting plants and is used among other things to "clean" soils from weeds.
So if you want to use leftover fertilizer for its intended purpose make sure to read up on how to use it properly. Applied at the wrong time it may do
more harm than good.
It should also be noted that cyanamide is somewhat toxic and might inhibt acetaldehyde dehydrogenase, which could lead to amplified hangover effects
if alcohol was consumed.
Pictures:
Commercial calcium cyanamide fertilizer. It is often black since the reaction by which it is made from calcium carbide gives carbon as a side product.

This is the shape dicyandiamide takes when crystallizing after the synthesis:
  
After a recrystallization the crystal shape becomes this:
 
The copper guanylurea complex, left precipitated with ammonia, right precipitated with NaOH solution:

Sources:
The main source is [6] where I found the procedure.
[1] J. Haag, (1862). Ueber Dicyandiamid und eine neue daraus entstehende Base. Annalen Der Chemie Und Pharmacie, 122(1), 22–33.
doi:10.1002/jlac.18621220103
[2] Gmelins Handbuch der Anorganischen Chemie - Kohlenstoff Teil D 1, 8. Auflage, Weinheim/Bergstraße 1971, p. 280, 282, 292f.
[3] https://de.wikipedia.org/wiki/Metformin
[4] J. B. Moffat, (1983). The dimers of cyanamide. Journal of Molecular Structure: THEOCHEM, 94(3-4), 261–265. doi:10.1016/0166-1280(83)80133-x
[5] Total synthesis of metformin, https://www.sciencesnail.com/science/total-synthesis-of-metf...
[6] J. Söll, A. Stutzer, (1909). Mitteilungen über einige neue Verbindungen, die aus Guanylharnstoff und aus Diguanid erhalten wurden. Berichte Der
Deutschen Chemischen Gesellschaft, 42(4), 4532–4541. doi:10.1002/cber.19090420453
[7] W. L. F. Armarego, Purification of Laboratory Chemicals 6th Edition 2009, p. 119
[8] Y. Sankaranarayanan, H. K. Sen, Manufacture of urea, dicyandiamide, melamine, guanidine carbonate and guanyl urea sulfate, 1943. https://insa.nic.in/writereaddata/UpLoadedFiles/PINSA/Vol09_...
[9] Ludwig F. Audrieth, Inorganic Synthesis Vol. III, 1950, p. 43f.
[10] Beilsteins Handbuch der Organischen Chemie Dritter Band - Acyclische Oxycarbonsäuren und Oxocarbonsäuren, Vierte Auflage Berlin 1921, p. 91ff.
For the german readers, I have written a more detailed writeup in german under https://illumina-chemie.de/viewtopic.php?p=81117#p81117
[Edited on 12-3-2021 by Diachrynic]
we apologize for the inconvenience
|
|
|
Alkoholvergiftung
Hazard to Others
Posts: 163
Registered: 12-7-2018
Member Is Offline
|
|
Old patents says if you add ammonia the reaction is faster and the yield is higher. Ive plan to when the weather is better an cold extraction on one
part calciumcyanamid 5 parts water. You should get cyanamid solution. If you add than ammonia it forms dicyandiamid.
Other way if you need cyanamid you can mix the calciumcynamid with oxalic acid and add slowly water. So its pure.(foot note)
|
|
|
Boffis
International Hazard
Posts: 1852
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Nice work Diachrynic! I have been meaning to do a write up on dicyandiamide for some time. I have prepared a good deal of dicyandiamide by this route
over the years (I purchased a 25kg bag of the stuff from an agricultural store). I have tried various modification including adding ammonium sulphate
to reduce the solubility of the Ca(OH)2, the liberated ammonia raising the pH and promoting the dimerisation of the cyanamide but it didn't seem to
change thing and I invariably got similar yield to your except on a few occasions when I got a lot of urea instead. I also tried making free cyanamide
but this was less successful.
Dicyandiamide is the starting material for many interesting compounds.
|
|
|
Engager
Hazard to Others
Posts: 295
Registered: 8-1-2006
Location: Moscow, Russia
Member Is Offline
Mood: Lagrangian
|
|
Dicyandiamide can also be used to make guanazole, precursor for triazole compounds.
Preparation of 3,5-diamino-1,2,4-triazole (guanazole) from dicyandiamide

Prepare solution of 105g hydrazine dihydrochloride (1 mol) in 250 ml of water (Note #1). Solution is placed into
beaker fitted with thermometer for measuring temperature of the liquid, mechanical stirrer and is placed on heating water bath. Solution is heated to
60oC and 84g of dicyandiamide (1 mol) is added in small portions with constant stirring (Note #2). After addition of last portion of dicyandiamide
reaction mixture is allowed to sit for 2 hours at 60-80oC (Note #3). Produced transparent solution of guanazole is cooled down to room temperature and
379 ml of 40% nitric acid is added (3 mol, 192 ml commercial 70% HNO3 + 202 ml of water). Reaction mixture is cooled to 0C on ice bath to affect
precipitation of guanazole nitrate (Note #4). Precipitate is separated on Buchner funnel and recrystallized from 165 ml of hot water (below 65oC !!!),
yielding 135g of guanazole nitrate mixture (Note #5), 72% of theory. Free guanazole can be obtained by neutralization of nitrate salt with alkali,
followed by crystallization. White crystalline solid with m.p. 204oC (with decomposition), soluble in water, forms mono and dibasic salts.
Note #1. Reaction only proceeds well in strongly acidic conditions, those are provided by
hydrazine dihydrochloride and allow protonation of cyano group in dicyandiamide, as the first stage of reaction’s mechanism. Chloride anions present
are known as good promoter of additions to the cyano group and allows high selectivity towards guanazole. Usage of hydrazine monohydrochloride or
sulphate dramatically reduces reaction yield.
Note #2. Reaction proceeds with notable evolution of heat, while temperature of the process should
not exceed 50-60oC due to formation of notable quantities of byproducts, like 3,5,7-triamino-1,2,4-triazolo[4,3a]-1,3,5 triazine, yield of the later
reaches 10-13% at 100oC. Dicyandiamide should be added in portions to control exotherm and to prevent formation of undesirable byproducts.
Note #3. Solution of hydrazine dihydrochloride has notable yellow color, during reaction process it
diminishes to the golden color of guanazole salt solution. Reaction greatly speeds up at higher temperatures, at 80-100oC it completes in several
minutes, but to reach maximum selectivity and to minimize amount of byproducts it’s better to carry out whole process at 50-60oC in duration of 2
hours.
Note #4. Solubility of guanazole dinitrate strongly depends on temperature, and is illustrated by
following numbers (g in 100 ml): 230 (80оC), 158 (70оC), 112 (60оC), 52.4 (40оC), 14.7 (20оC), 5.9 (5оC). Such solubility curve allows
separation of guanazole in form of nitrate salt, bypassing normal process requirements (evaporation of solvent in vacuum followed by extraction). Used
amount of nitric acid is optimal for separation of maximum amount of salt. Guanazole dinitrate salt is unstable on storage on the air and slowly loses
nitric acid.
Note #5. Warning! Attempt of determination of product melting point in thin capillary led to
explosion, that occurred before melting (at ~120-130oC) and led to destruction of thermometer. Titration of product with NaOH showed, that product
contains 1.4 mol of HNO3 for 1 mole guanazole and is mixture of mono and di nitrates of guanazole. According to reference data, stability of dinitrate
is limited by 90oC, leading to decomposition of compound, that caused explosion mentioned above.
Qualitative test for guanazole. Copper (II) sulfate pentahydrate is mixed with same visual amount
of sodium acetate and the mixture is dissolved in minimum amount of water, leading to deep blue solution, containing copper (II) acetate; when few
drops of the reagent are added to neutral solution of guanazole, light green precipitate is immediately formed. According to reference data its
composition is DAT*CuSO4*H2O. Addition of additional quantity of reagent can lead to dissolution of precipitate with formation of dark green solution
containing copper-DAT multinuclear complexes. Reaction of hydrazine salt with same reagent leads to markedly different process: at first solution
turns blue/violet and either changes color to opaque yellowish with quick liberation of nitrogen and precipitation of metallic copper, or deposits
white insoluble precipitate containing copper (I) compound.
Attachment: US2648671 PREPARATION OF GUANAZOLE.pdf (84kB)
This file has been downloaded 302 times
Attachment: JCS page 517 guanazole.pdf (215kB)
This file has been downloaded 303 times
Attachment: Synthesis of Cobalt(III) Ammine Complexes as Explosives for Safe Priming Charges (Tselinsky 2003).pdf (41kB)
This file has been downloaded 272 times
Attachment: Synthesis and Characterization of 3,5-Diamino-1,2,4-triazolium Dinitramide.pdf (675kB)
This file has been downloaded 359 times
Attachment: Guanazole copper complexes (aznar2006).pdf (218kB)
This file has been downloaded 328 times
[Edited on 16-3-2021 by Engager]
|
|
|
Engager
Hazard to Others
Posts: 295
Registered: 8-1-2006
Location: Moscow, Russia
Member Is Offline
Mood: Lagrangian
|
|
Preparation of 3,5-dinitro-1,2,4-triazole from guanazole

To the 300 ml reaction beaker, equipped with dropping funnel and efficient mechanical stirrer (Note #1), placed 55 g
of sodium nitrite (0.8 mol), 90 ml water and mixture is heated on water bath at 50oC until solid dissolves. Dropping funnel is filled with solution
10g of 3,5-diamino-1,2,4-triazole (0.101 mol), 125 ml water and 9 ml of concentrated sulfuric acid (0.168 mol, Note #2). Solution of dropping funnel
is added drop wise to vigorously stirred solution of sodium nitrite, while maintaining temperature of 50oC at such rate, that whole addition process
take around 4 hours. After each hour of addition mixture is allowed to sit for 15 minutes at 50oC before addition is resumed again (Note #3). After
addition of each drop yellow foam forms around the drop point, that quickly dissolves in reaction mixture (3 mono and 3,5 dinitrosoamino-1,2,4
triazoles) and some gasses are formed (nitrogen oxides are easily detectable by odor); while at places where released gasses contact with solution of
guanazole in addition funnel intense red insoluble solid forms (Note #4). At the beginning of the addition process, reaction mixture is transparent
and has lime-yellow color, at the middle of addition it turns into tea color (Danger!!! Note #5). After complete addition of guanazole, stirred
reaction mixture is kept at 50oC for 1 hour, when temperature is raised to 60oC and solution is stirred for 1 more hour (Note #6). After aging
process, with vigorous stirring 22.7g of 50% sulfuric acid (0.232 mol) is added dropwise during 1-2 hours, on completion mixture is neutralized by
baking soda and stirred at boiling water bath for 1 hour (Note #8). Reaction mixture from small amount of colorless solid impurity (polytriazenes) and
allowed to evaporate slowly at room temperature (Note #9). Crude mixture of salts (consisted mostly of Na2SO4*10H2O, NaDNT and some NaNO3) is
extracted by 150 ml of the acetone (150 ml), filtered from insoluble material, 15 ml of water is added and extract is allowed to evaporate slowly at
room temperature (Note #10). Sodium salt of 3,5-dinitro-1,2,4-triazole shows some isomorphism with sodium sulfate, that leads to occlusion of product
in crystals of sodium sulphate, so for more complete isolation of product acetone extraction is repeated 2-3 more times, with though grinding of
solids before each extraction (Note #11). After 3-x extraction by acetone 10.2g (64% of theory, ref [4] gives 66% yield) of sodium salt of
3,5-dinitro-1,2,4-triazole is collected in form of red-orange crystalline material (Note #12). Separation of free DNT from the sodium salt was
described in refs [3,4].
Note #1. According to data from ref [4] yield and purity of dinitrotriazole are improved when
mechanical stirring was used instead of magnetic one, and also on slower addition of guanazole solution.
Note #2. Selection of sulfuric acid (instead of HCl or HNO3, often used for such reactions) is
based on several reasons. First one is its inability to enter the redox processes (direct substitution of diazonium group require electron transfer
from anion), leading to higher purity of product than in case of HCl (in later case product is contaminated by chloro-triazoles). Second is that
it’s diazonium salts are more stable than extremely sensitive and unstable diazonium nitrates, and precipitation of relatively low soluble guanazole
nitrate salts is the feeding line is prevented.
Note #3. Employed synthetic procedure is different to normal Sandmeyer diazotizations preformed in
presence of copper salt catalysts at low temperature and is instead the chain radical process driven by Srn1 mechanism [1,2,3]. Possibility of direct
substitution of diazonium group by this mechanism is based on the fact that redox potentials of nitrite ion is close to the classical Cu(2+)/Cu(+)
redox pair, that provide electron transfer in Sandmeyer and Gatterman’s reactions. Formation of diazonium salt is fast process already at 0oC, while
process of replacement of diazonium group with nitrite ion is much more slow and proceeds readily only at temperatures around 50oC. So, the method
described above, allows to combine diazotization and replacement processes at the same time, allowing to mitigate accumulation of highly dangerous and
unstable diazonium intermediates, that are present at the reaction mixture. Slow addition rate of guanazole solution and 15-minute cooldowns serve the
same purpose.
Note #4. Nature of this product is not completely clear, it’s insoluble in water and is probably
a triazene formed by azo-coupling of diazonium salt (formed by reaction of escaping nitrous gases with guanazole solution) with pristine molecules of
guanazole. Authors of work [5] describe several coupling reactions of guanazole, and from visuals and properties product in question corresponds to
compound IV – 3,5-bis{[3-(5-amino-1,2,4-triazolyl)]triazenyl}-1,2,4-triazole. Under the microscope compound appears as small orange-red crystals,
having high affinity for water. Taking to account visual similarity it’s still unclear, why exactly this products form or it is something else
entirely. Whatever is the case, formation of this product is undesirable as it will lead to contamination of target compound, while diazotization out
of boundary of main reaction solution leads to danger of crystallization and explosion of formed diazonium salts, so effort should be taken to
minimize contact between escaping nitrous gases with guanazole solution in addition funnel.
Note #5. DANGER! Approximately at the middle of addition period, strong micro-detonation occurred,
accompanied by visible white flash, this was without a doubt the decomposition of tiny amount of unstable diazonium compound. It is well known that
stability of diazonium salts greatly depends on anion and composition of aromatic substrate: electron-donating groups in the ring and voluminous
non-oxidizing anions like tetrafluoroborate or sulfate provide stabilizing action, while electron-withdrawing groups in the core greatly destabilize
diazonium salts. Guanazole contains two amino-groups; those can be diazotized or be replaced by nitro group in the same time leading to formation of
extremely unstable and dangerous diazonium salts rivaling, instability of the 5-diazotetrazole. Inspection of damage shown explosion occurred on the
surface of mechanical stirrer and was probably caused by contact of drops of guanazole solution with nitrous gasses, followed by drying the stream of
air at the stirrer rod’s surface. Event again places emphasis on importance of safety measures and equipment, that should be employed whole time and
especially during guanazole addition process.

Note #6. Purpose of the aging process is to maximize substitution completion and decomposition of
unstable diazonium intermediates. After complete addition solution was transparent, had color of dark tea (with some reddish tinge), and contained
only small trace of crème-white precipitate (polytriazene).
Note #7. Addition of sulfuric acid induces sizeable form formation, caused by formation of nitrogen
oxides, originating from decomposition of excess of sodium nitrite. After addition of most acid release of gasses and foaming are stopped, mixture was
fully transparent, had lime-yellow color and show pH <= 1.
Note #8. Reaction equations show, that synthetic procedure should spent 3*0.1*0.5 = 0.15 mol of
[H+], while 0.4 was added in total, so neutralization should require 0.25 mol of hydrocarbonate (21g). In reality neutralization took 19.4g, and
caused color change to tea color, formation of trace of insoluble precipitate, separated by filtration through the cloth, pH = 7-8.
Note #9. Literature data on DNT salts [6] suggest high sensitivity of anhydrous salts. Slow
evaporation at room temperature insures formation of safe, hydrated product. Evaporation to dryness on 4 plates at room temperature took around 3
days. Examination of product under microscope shown large amount of rod habit crystals (sodium sulfate octahydrate) and yellow DNT salt in form of
plates and needles. After complete drying some small amount of impurity is seen in from of star like agglomerates of orange-red color.
Note #10. Intentional addition of water is aimed on separation of product in the form of dyhydrate.
Fast evaporation of sample of acetone solution leads to formation of pike shaped golden crystals of product observed under the microscope. According
to reference [6] anhydrous salt is colorless, while hydrate is yellow-orange. Material obtained on evaporation of solvent with added bit of water is
orange-yellow microcrystals.
Note #11. Part of allotropic forms of hydrates of Na2SO4 and the anhydrous salt can form monoclinic
crystals showing some isomorphism with monoclinic crystals of NaDNT and it’s dehydrate, that leads to easy occlusion of DNT salt by hydrated sodium
sulfate crystals. Yield after one extraction is only 35% from theory, so extraction is conducted multiple times. Major part of solid residue are
Na2SO4*10H2O (dec.temp. 35oC), Na2SO4*7H2O (dec.temp. 25oC) with some amount of anhydrous salt. Sodium sulfate have anomalous solubility curve below
40oC (during cooling from 25oC to 0oC solubility is reduced 10 times and is around 5g of salt in 100 ml at 0oC) that can be used to separate most
sodium sulfate before the next extraction. Solution of this salt have notable habit for form supersaturated solutions, that suddenly crystalize as a
whole on lowering the temperature and have extremely fluffy structure and are hard to filter. So it’s advantageous to perform cooling from 40oC to
0oC very slowly, adding some seed crystal, that leads to formation of huge (several centimeter long) crystals of sodium sulfates that occlude minimal
amount of mother liquor and DNT salt. For more complete separation of the later, solid residue from first extraction is ground to fine powder and
extracted by new portion of acetone, undissolved solid is dissolved in minimal amount of boiling water (around 150 ml) cooled to 40oC, seed crystal is
added and solution is slowly cooled to 0C, precipitated crystals of Na2SO4*10H2O are separated by filtering and mother liquor is evaporated at room
temperature, grounded and extracted by acetone the third time. Yield after first extraction is around 30%, second extraction gives 25% more, and third
one gives more 10%. Total yield reaches 10.2g of NaDNT (64% of theory).
Note #12. Crystallization from anhydrous acetone and from acetone with addition of water leads to
product containing some minor amount of (like 1-2%) of ruby-red crystals clearly visible under microscope. This impurity is highly soluble, since it
forms on the outer edges of NaDNT crystals. Nature of this impurity is unknown.
References:
[1] Aromatic substitution by the SRN1 mechanism; Joseph F. Bunnett; Accounts of Chemical Research 1978 11 (11), 413-420; DOI: 10.1021/ar50131a003
[2] Radical nucleophilic substitution mechanism in the reactions of arenediazonium cations with nitrite ion; P.R.Singh, Ramesh Kumar, R.K.Khanna;
Tetrahedron Letters Volume 23, Issue 49, 1982, Pages 5191-5194; DOI: 10.1016/S0040-4039(00)85794-9
[3] Heterocyclic nitro compounds. I. Synthesis of nitro derivatives of 1,2,4-triazole, 1,3,4-thiadiazole, tetrazole, 1,3,4-oxadiazole, and pyrazole by
the noncatalytic replacement of the diazo group by the nitro group ; L.I. Bagal, M.S. Pevzner, A.N. Frolov & N.I. Sheludyakova; Chemistry of
Heterocyclic Compounds volume 6, pages 240–244 (1970) ; DOI: 10.1007/BF00475005
[4] Preparation and characterization of 3,5-dinitro-1H-1,2,4-triazole ; Haiges, R. and Bélanger-Chabot, G. and Kaplan, S. M. and Christe, K. O.;
Dalton Trans. V44, I.16, P 7586-7594 (2015); DOI: 10.1039/C5DT00888C
[5] The Diazotization and Autocoupling of Guanazole; M. Hauser; The Journal of Organic Chemistry 1964 29 (11), 3449-3450; DOI: 10.1021/jo01034a532
[6] Synthesis and structural characterization of 3,5-dinitro-1,2,4-triazolates; R. Haiges, G. Bélanger-Chabot, S. M. Kaplan, and K. O. Christe;
Dalton Trans. 2015 Feb 21;44(7):2978-88; DOI: 10.1039/c4dt03583f.




Attachment: [1] Aromatic Substitution by the SRN1 Mechanism (bunnett1978).pdf (1.1MB)
This file has been downloaded 347 times
Attachment: [2] Non Cat Radical Sanmeyer - Srn1 with nitrites !!! (singh1982).pdf (258kB)
This file has been downloaded 271 times
Attachment: [3] bagal1970.pdf (361kB)
This file has been downloaded 278 times
Attachment: [4] Preparation and characterization of 3,5-dinitro1H-1,2,4-triazole (haiges2015).pdf (1.5MB)
This file has been downloaded 360 times
Attachment: [5] The Diazotization and Autocoupling of Guanazole - hauser1964.pdf (282kB)
This file has been downloaded 278 times
Attachment: [6] Synthesis and structural characterization of 3,5-dinitro-1,2,4-triazolates (Salts).pdf (1.7MB)
This file has been downloaded 288 times
[Edited on 16-3-2021 by Engager]
|
|
|
Boffis
International Hazard
Posts: 1852
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
@Diachrynic; a question for you. When you use the raw pellets of calcium cyanamide without crushing them do you find that they disintegrate during the
boiling? It just surprises me that 30 minutes boiling can extract the cyanamide without disintegrating the pellets completely.
When I tried this before I crushed the pellets until they would all pass a 0.5mm sieve. When I tried it with whole pellets I added sulphuric acid to
see if it would help as per your ref. 8 above. The pellets were still slow to disintegrate and longer boiling resulted in much urea. I tried using
ammonium sulphate to prevent the reaction mixture from becoming too acid while precipitating the calcium but I still only got about 11g per 100g of
cyanamide.
Oh and welcome back Engager! Where have you been hiding for the last few years? Clearly doing some very interesting work on 1,2,4-triazole
derivatives, very nice! 
[Edited on 16-3-2021 by Boffis]
|
|
|
Diachrynic
Hazard to Others
Posts: 222
Registered: 23-9-2017
Location: western spiral arm of the galaxy
Member Is Offline
Mood: zenosyne
|
|
Engager, fantastic work on the triazoles! I did not expect you to post today. A shame the guanazole seems to require hydrazine
dihydrochloride, well, I guess a double displacement from the sulfate it is. Maybe I will give this a go, now that I have access to dicyandiamide per
this process.
Quote: Originally posted by Boffis  | | @Diachrynic; a question for you. When you use the raw pellets of calcium cyanamide without crushing them do you find that they disintegrate during the
boiling? It just surprises me that 30 minutes boiling can extract the cyanamide without disintegrating the pellets completely. |
That is exactly what I thought which is why I started the experiments with pulverized fertilizer. When using the pellets as is - it's hard to tell how
many disintegrate, if I had to estimate maybe 40% do. Most of them are still distinctly prill shaped. To my surprise it had no effect on the yield
whatsoever. I am not sure why, but it works I guess, so I don't complain.
I find it weird that adding sulfuric acid doesn't seem to help the yield that much. Maybe the control for that to matter just has to be more precise,
but I checked with pH paper and hit the window pretty good I think. I also wonder where the other 50% of the yield go. Evidently, not into the product
as impurity, because melting point and etc. all match. It also can't go to ammonia, because there was only a very slight smell of that. I did it
indoors and if half of the dicyandiamide would have decomposed to ammonia I would have known.
we apologize for the inconvenience
|
|
|
Boffis
International Hazard
Posts: 1852
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Hi Diachrynic, I was interested in your water only hydrolysis of calcium cyanamide and since I have lots of it I thought I would give it a go. The
results were pretty much as you described above. In one scaled-up experiment after washing the leached residue I washed the residue through a
stainless steel sieve (about 500micron apps.) and then added the coarse material back to the liquor after filtering off the crystals of dicyandiamide
and repeated the extraction a second time. I recovered an extra 3.6g from 425g of starting Ca cyanamide. The initial crop of crystals was 26.0g so in
spite of the fact that the prills didn't completely disintegrate there is very little cyanamide left in the residue from a single extraction. The wash
water from the first pass was not added to the second extraction and deposited an additional 5.1g of dicyandiamide on standing overnight. From this I
conclude that careful handling of the washings is more important and useful than trying to extract more dicyandiamide from the residue.
This experiment was carried out on material from a sack which has been open for 5 years and has started to turn dusty grey so these yields are not
that bad. I carried out the preparation experiment using material which had been transferred to air-tight plastic buckets.
I also tried evaporating down the mother liquor left after recovery of the dicyanamide crystals but this deposited much calcium carbonate and turned
green. I filtered of the solids but the liquor rapidly produced more ppt and became quite viscous on cooling. Nothing useful crystallised from it.
Sodium bicarbonate can be used to precipitate the excess calcium before evaporation but this didn't improve things at all.
I have previous experimented with the preparation of guanylurea nitrate and other derivatives from dicyandiamide in this thread:
http://www.sciencemadness.org/talk/viewthread.php?tid=149987...
|
|
|
Illegal Parkinson
Hazard to Self
Posts: 80
Registered: 2-10-2005
Member Is Offline
Mood: No Mood
|
|
Dear Sir, I have been interested in these synthesis myself.
https://onlinelibrary.wiley.com/doi/book/10.1002/97804701323...
The reference you need is L. A. Pinck R. L. Phelps.
A quantitative yield of dicyanodiamide was obtained by the authors in this laboratory practical.
|
|
|
Boffis
International Hazard
Posts: 1852
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
@ Illegal Parkinson, I am aware of this published preparation of dicyandiamide but you are missing the subtlety of the synthesis, the problem is not
converting cyanamide into its dimer, it is obtaining the cyanamide in the first place. The isolation of the cyanamide is the tedious bit. The beauty
of Diachrynic's procedure is that it bypasses the isolation of the later, it is very quick and simple and the required materials are very cheap and
available.
The solubility of cyanamide is about 70g /100ml and it has a strong tendency to both polymerise and to hydrolyse so isolation is difficult. You need
to read and consider the practical application of the InorgSynth vIII previous section on the preparation of cyanamide, it is exceedingly tedious. If
cyanamide is your desired goal there are better methods such as the acetic acid disintegration of the calcium cyanamide followed by ether extraction
but this is a hell of a lot of extra work if dicyandiamide is your desired product.
|
|
|
Engager
Hazard to Others
Posts: 295
Registered: 8-1-2006
Location: Moscow, Russia
Member Is Offline
Mood: Lagrangian
|
|
| Quote: Originally posted by Diachrynic | | I find it weird that adding sulfuric acid doesn't seem to help the yield that much. Maybe the control for that to matter just has to be more precise,
but I checked with pH paper and hit the window pretty good I think. I also wonder where the other 50% of the yield go. Evidently, not into the product
as impurity, because melting point and etc. all match. It also can't go to ammonia, because there was only a very slight smell of that. I did it
indoors and if half of the dicyandiamide would have decomposed to ammonia I would have known. |
Several thought why this is the case.
Have you considered how many cyanamide nitrogen your fertilizer really contain? Did you made tests to determine this content? How
old is your fertilizer and how it was stored? (It's well known that while storing on the air calcium cyanamide reacts slowly forming ammonia: CaNCN +
3H2O => CaCO3 + 2NH3) How you can even judge on yield if you don't know exact cyanamide content in your source material? You believe specification
digits?
Dicyandiamide is itself unstable towards hydrolysis. In acid medium it attaches water forming guanylurea, this can hydrolyze further forming
guanidine. Hydrolysis by aqueous ammonia proceeds in same way, forming guanylurea first, it is when converted to guanidine carbonate, and this later
can enter secondary reaction with ammonia forming melamine. Hydrolysis by bases like NaOH leads to same products, guanylurea and guanidine. Formation
of guanylurea is fast, while it's conversion to guanidine is way slower and takes more effort. Please take note that formation of guanylurea
does not lead to formation of NH3 or visible gas evolution, it's addition reaction on nitrile group, so you don't have visual indication of this
process. NH3 and CO2 are only formed on very slow hydrolysis of guanylurea to guanidine. Had you tried to find guanylurea in the mother
liquor after product separation?
Do you know that dicyandiamide is soluble in alkali and can even form solid salts that can be prepared in water solutions. Check out patent
US2357261A. Pure diacyandiamide is neutral in solution, your reaction mass is alkaline, but i do not see any acid adition in your procedure.
Had you considered the possibility of incomplete polymerisation of cyanamide to dicyandiamide? Process can be fairly slow and depends strongly
on pH of solution. Had you tried to perform test for cyanamide presense in mother liquor after separation of your product?
Some read attached.
Attachment: Dicyandiamide hydrolysis.pdf (633kB)
This file has been downloaded 769 times
Attachment: The constitution of carbamides. Part II. The relation of cyanamide to urea.pdf (231kB)
This file has been downloaded 280 times
[Edited on 23-3-2021 by Engager]
|
|
|
Boffis
International Hazard
Posts: 1852
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
@Engager, I notice when looking closely at the mechanism for the formation of guanazole you have given above that ammonia is formed in the form of
ammonium ions. Would it be possible to use a hydroxylammonium salt in place of the hydrazine salt in which case water and H+ would be the byproducts?
|
|
|
Engager
Hazard to Others
Posts: 295
Registered: 8-1-2006
Location: Moscow, Russia
Member Is Offline
Mood: Lagrangian
|
|
| Quote: Originally posted by Boffis | | @Engager, I notice when looking closely at the mechanism for the formation of guanazole you have given above that ammonia is formed in the form of
ammonium ions. Would it be possible to use a hydroxylammonium salt in place of the hydrazine salt in which case water and H+ would be the byproducts?
|
I had not seen reaction of dicyandiamide with hydroxylamine anywhere in references. If you check out structure of intermediate performing ring closure
it's possible what addition product with hydroxylamine can close the ring forming 3,5-diamino-1,2,4-oxadiazole instead, or it can split water with
formation of biguanide. Hydroxylamine general reactivity is somewhat different from hydrazine, and analogy can fail at any stage, however i believe,
that such kind of reaction is possible.
[Edited on 28-3-2021 by Engager]
|
|
|
Engager
Hazard to Others
Posts: 295
Registered: 8-1-2006
Location: Moscow, Russia
Member Is Offline
Mood: Lagrangian
|
|
It may be interesting, for some, that similar process with ammonium chloride in the melt at certain temperatures can produce biguanidine and guanidine
salts. Here is part of my lab book:
Experiments with dicyandiamide/ammonium chloride melt. Production of guaindine and biguanide
salts.

Experiment №1. 25.2g of dicyandiamide and 40.1g of ammonium
chloride were thoroughly grounded and mixed together and placed into iron can from the juice with top cut open (volume was approximately 240 ml). Can
was placed into stand holder and was heated by the open flame of gas oven. No visual change was observed during first 10 minutes, some white fumes of
sublimed ammonium chloride were notable. Temperature of mixture was observed during stirring, by means of the thermocouple imbedded into glass
stirring rod. After temperature reached 160oC, layer of liquid melt was observed at the places of contact of mixture with iron can surface (where heat
transfer is better), after this big exotherm sets up with almost immanent rise of temperature to 230C, after this whole mixture was liquefied into
mobile liquid. After exothermic reaction started heating was discontinued and mixture was left to cool down, causing solidification of reaction mass.
Solid product was exceptionally hard and could be hardly broken by strikes of hammer. Solid mass was dissolved in 150 ml of boiling water, leading to
almost complete solution with almost no insoluble material (almost complete absence of ameline). Separation of solid impurities by filtering led to
clear transparent solution with slight yellowish tinge. Next, solution of copper (II) sulfate in concentrated ammonia was added until solution
achieves blue coloration (excess of copper tetramine sulfate). Additions of first few drops of solution led to formation of reddish-crème coloration,
that was almost immediately discharged and changed into dark blue. Filtering afforded 0.5g of brownish-red crystals of unknown composition. Absence of
copious amount of precipitate with copper (II) tetramino sulphate, shown almost complete absence of biguanide in reaction mixture.
Target compound was not separated, while repeating above procedure, given in reference [1]. Details of method from
reference are however uncertain due to inability to replicate heating conditions of Bunsen burner flame, depending from gas flow and heat transfer
conditions. Investigation of literature data shown, that strong heating (even in small duration) what took place in the experiment (around 180oC)
leads to guanidine with only trace quantity of biguanide, while stronger and longer heating leads to ameline as major reaction product. No attempt was
made to separate the guanidine product, however it was clear a major product of above experiment and should have formed in nearly quantitative yield.
It was decided to repeat the synthesis by using careful heating on liquid petroleum paraffin bath.
Experiment №2. 25.2g of dicyandiamide and 40.1g of ammonium
chloride were thoroughly grounded and mixed together and placed into iron can from the juice with top cut open (volume was approximately 240 ml). Can
was placed into stand holder and was heated by molten petroleum paraffin heating bath. When heating bath temperature of 170oC was reached, heating was
discontinues and resumed only when temperature dropped below 150oC. In those conditions petroleum paraffin is somewhat volatile and produces some
white smoke and is readily detectable by odor, along with some mild odor coming from reaction mass. Heating in bath at 150-170oC for 30-40 minutes had
not produced noticeable changes in the reaction mixture, no liquid melt areas were seen. After approximately 1 hour of heating some liquid areas began
to form, after 1.5 hours whole mixture was melted, forming viscous liquid similar to honey in consistency. After complete melting mixture was heated
to 150-170oC for 15 more minutes, during this time viscosity dropped and mixture became easier to stir. Heating was discontinued and liquid melt was
poured onto paper and was grounded before full solidification. Resulting mass was dissolved in 150 ml of boiling water, most of solid dissolved, but
some small amount of ameline residue was also present (weighted around 3g). Filtrate was completely colorless solution, that on addition of ammonical
copper (II) tetramino sulfate immediately produced huge quantity of pink precipitate, solution remained completely colorless after addition of each
portion, before the excess of the reagent was added, leading to deep blue coloration of solution. Full precipitation took about a half of theoretical
amount of ammonical copper (II) tetramine sulfate. Filtering afforded copper biguanide sulfate in fair yield as rose-red solid, what is washed with
water and dissolved in 35 ml of 10% sulfuric acid (the temperature should not exceed 90°). The resulting solution is then cooled by immersion in an
ice bath. The crude crystals which form are separated and dissolved in 25 ml. of boiling water and the solution is cooled in ice. Again the crystals
which form are dissolved in 25 ml. of boiling water and the solution is cooled in ice. The resulting colorless crystals of biguanide sulfate 2-hydrate
are filtered, washed first with 10 ml. of cold water and then with 10 ml. of absolute ethanol, and dried at 110° for about 15 hours. The yield of
anhydrous biguanide sulfate is 10.1 g. (16.9%); melting point 232oC.
Properties: Pure anhydrous biguanide sulfate is a white
amorphous solid that is not very soluble in cold water but is soluble in hot water, from which it crystallizes in colorless glistening rhombic
crystals. An aqueous solution of biguanide sulfate is acidic to litmus. The compound does not dissolve in absolute ethanol to any appreciable extent
and is insoluble in common organic solvents. Reaction of compound with theoretical amount of NaOH in dry methanol allows separation of pure biguanide
free base, with 90% yield, m.p. 134oC (decomposition at 138oC).
Refs:
[1] Biguanide Sulfate ; Inorganic Syntheses ; In book: Inorganic Syntheses, Volume 7 (pp.55-58) ; DOI: 10.1002/9780470132388.ch14
[Edited on 28-3-2021 by Engager]
|
|
|
Boffis
International Hazard
Posts: 1852
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Yes I would anticipate the initial condensation product to be guanyl formamidoxime (N-hydroxybiguanide) but I would then expect it to cyclotize. The
question then is; does it cyclotize by the elimination of water or ammonia?
I can't see how the amidoxime group can eliminate water to form biguanide though. The condensation of ammonia with dicyandiamide gives biguanide
briefly on route to guanidine.
Ah; you managed to post this while I was responding to you earlier post .
That's a good yield you got there, I only got 3.6% with this procedure. I also tried the ammonium iodide fusion route but while it is supposed to give
a much better yield than ammonium chloride the work-up is very messy. I also tried ammonium nitrate, this gave me my best yield of biguanide at just
under 10%. This is why I have decided to try the O-methyl isourea route from cyanamide. I am currently experiemnting with the isolation of cyanamide
from crude calcium cyanamide.
[Edited on 28-3-2021 by Boffis]
|
|
|
Engager
Hazard to Others
Posts: 295
Registered: 8-1-2006
Location: Moscow, Russia
Member Is Offline
Mood: Lagrangian
|
|
| Quote: Originally posted by Boffis | | That's a good yield you got there, I only got 3.6% with this procedure. I also tried the ammonium iodide fusion route but while it is supposed to
give a much better yield than ammonium chloride the work-up is very messy. I also tried ammonium nitrate, this gave me my best yield of biguanide at
just under 10%. This is why I have decided to try the O-methyl isourea route from cyanamide. I am currently experiemnting with the isolation of
cyanamide from crude calcium cyanamide. [Edited on 28-3-2021 by Boffis] |
I did that biguanide experiment like 8 years ago or more. From what i remember and as it can be seen from experiment details: temperature control is
of critical importance for yield of material. If you succeed on this regard yields can be fair.
O-methyl isourea can be nice, i guess you are trying to react cyanamide with sulphuric acid + methanol, or you decide to go some other route? It's sad
though same scheme can't work on getting O-alkyl isosemicarbazide, that i wanted for the long time, to get my hands on some at least semi pure
diaminoguanidine. I was considering making some diethylcyanamide from diethylsulfate and CaNCN, and when reacting it with hydrazine, however i really
hate those organic sulfates, so i never tried.
I had prepared S-ethyl isothiosemicarbazide hydrobromide some long time ago to get the diaminoguanidine from it. However i immediately noticed strong
red color of the solid on contact with air due to the presence of tetrazine compound and decided to save the material for the future experiments with
tetrazine compounds. This stuff however is incredibly smelly so i never bothered to touch it anymore due to the "funny" odor of C2H5SH making
everything around smearing worse than the pile of s***...
Keep us posted about O-methyl isourea, it can be handy.
[Edited on 28-3-2021 by Engager]
|
|
|
Boffis
International Hazard
Posts: 1852
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
High Engager, Some time back I started trying to prepare diaminoguanidine by much the same process as you were envisaging from S-ethyl
isosemicarbazide hydrobromide but I decided to try butane bis(S-isosemicarbazide) dihydrobromide prepared from 1,4-dibromobutane and thiosemicarbazide
on the basis that the resulting butane-dimercaptol is much less volatile and hopefully less smelly than single mercaptols. I have tried this with
thiourea and dibromobutane to obtain aminoguanidine and the dimercaptol. It not only work but the dimercaptol is less smelly and smell more like
boiled cabbage or asafoeteda resin. I also tried thiourea and dibromoethane, this successfully gave ethylene bis(S-isothiourea) dihydrobromide in
excellent yield but on hydrazinolysis gave poor yields of anything identifiable. Interestingly 1,2 dichloroethane doesn't work with these reactions,
it seams that bromides (or possibly iodides) are required. Small scale experiments show that 1,6-dibromohexane reacts with both thiourea and
thiosemicarbazide so this might offer a route with even less horrid smelling byproducts .
This project stalled for want of time and I have been side-tracked since then but I will get back to this topic again soon.
I will let you know how I get on with the cyanamide prep. (Werner, TransCS., p1325, 1916), success here is key to the next stage to prepare the
O-alkylisoureas.
|
|
|
|









