| Pages:
1
2
3
4
5 |
Hey Buddy
Hazard to Others

Posts: 384
Registered: 3-11-2020
Location: Bushwhacker Country
Member Is Offline
|
|
I was hoping to get some advice. Im leaning towards this nickel complex but before I commit to getting into the habit of reduction of nitroguanidine
to aminoguanidium, I want to make sure there isnt a guanidium freebase complex equivalent. If there were say, Ni(Gu)3(ClO4)3 it could very well have
primary explosive capability but it would negate the need to reduce nitroguanidine which would simplify precursory steps. In this hope, I will try to
see if I can find something in guanidine. I have figured out the freebasing procedure as of tonight. GuNO3 is freebased in denatured alcohol by
heating to 68 C and adding stoich NaOH then filtering without wash. The freebase guaniidine disappears the moment water touches it.
I was hoping someone more familiar with the guanidine salts could help me with the next part. So far I plan to use metal chloride, then mix Ammonium
perchlorate/KClO4, (m)cl2, Gu in stochiometry for target compound. If KClO4 is used with chlorides then the KCL has low solubility in ethanol and
acetone. I have a huge collection of metals I could try, but Im not sure about what solvent to try reaction in, or if there would be a more logical
reagent? I can prepare anything, I would rather use a ClO4 salt as opposed to HClO4 because it is possible and simplified. If you have any ideas or
recommendations please advise. Im on standby until I can decide the solvent and I cant decide. SHould I just use water? I have no idea what target
complex solubility will be.
[Edited on 2-12-2022 by Hey Buddy]
|
|
|
Laboratory of Liptakov
International Hazard
Posts: 1339
Registered: 2-9-2014
Location: Technion Haifa
Member Is Offline
Mood: cool.gif
|
|
Certainly there are procedures (within the investigation of xy - guanidine) that no one has tried yet. Maybe you will be able to make something
useful, new, hitherto unknown. As the first in the world. But in that case, you also have to come up with a procedure. Also the first in the world.
I'm afraid your question is just that case. That is, how to simplify the procedure and achieve reliable DDT without the preparation of aminoguanidine.
(I repeat again: Amateur chemists are some of the most lazy scietist)
In exept of B(a)P
I consider myself one of the laziest researchers ever. That's why I invented Lithex. Which is a primary-secondary substance designed for the biggest
chemical slackers. Lithium perchlorate, hexamine and 5 drops of water are mixed in a pan. It evaporates at 175 Celsius. And substance is in the world.
[Edited on 2-12-2022 by Laboratory of Liptakov]
Development of primarily - secondary substances CHP (2015) Lithex (2022) Brightelite (2023) Nitrocelite (2024)
|
|
|
Hey Buddy
Hazard to Others
Posts: 384
Registered: 3-11-2020
Location: Bushwhacker Country
Member Is Offline
|
|
Ha, yes! But if there is a primary located in a simple guanidine, it will make everyone's life easier.
The zinc reduction of NQ is a mess and low yield where as the electro reduction on lead is much higher yield and cleaner, but requires the reduction
cell be made with a diaphragm and electolyte. So this step of reduction to aminoguanidine is a bottle neck. Likewise, if you go the other direction
and use hydrazine for ANQ, it is a bottleneck and the ANQ yield is relatively low with a good bit of material loss to diaminoguanidine etc.
Both directions, aminoguanidine <= and => ANQ have good primary explosive salts but take extra steps. Lazy chemists should be happy if there is
guanidine primary explosive complexes. It is likely there is.
Guanidine is freed from the nitrate in 10 minutes with NaOH in alcohol and then dries quickly. I have three batches from last night. Works great, low
losses. I will make some more and some metal perchlorates and waste some of it to try to find a good route. I will start with nickel perchlorate and
let you know which method works if a primary explosive is found that detonates unconfined like AGuNi salt.
|
|
|
B(a)P
International Hazard
Posts: 1116
Registered: 29-9-2019
Member Is Offline
Mood: Festive
|
|
LL not sure if I should be offended or not?
Hey Buddy check out the patent I referenced at the start of this thread. I think it has some info relevant to what you are trying to achieve.
|
|
|
Laboratory of Liptakov
International Hazard
Posts: 1339
Registered: 2-9-2014
Location: Technion Haifa
Member Is Offline
Mood: cool.gif
|
|
Hey Buddy, great info, thanks.....Guanidine free base is possible drying and storage...?...Good message. I am curious on any result on this field.
B(a)P.....Your holes in metal are excelent...Howgh...said Sitting Bull.
Development of primarily - secondary substances CHP (2015) Lithex (2022) Brightelite (2023) Nitrocelite (2024)
|
|
|
Hey Buddy
Hazard to Others
Posts: 384
Registered: 3-11-2020
Location: Bushwhacker Country
Member Is Offline
|
|
I will make a new thread about this in pursuit of guanidium complexes from the freebase so I dont drown B(a)Ps thread with offtopic.
BaP thank you for patent advice.
|
|
|
Hey Buddy
Hazard to Others
Posts: 384
Registered: 3-11-2020
Location: Bushwhacker Country
Member Is Offline
|
|
Ni(ClO4)2*6H2O from NiSO4 + 2KClO4
I've been working on this complex recently. I have prepared Ni(ClO4)2*6H20 via double decomposition from 2KClO4 + NiSO4 heated in dH2O until
dissolved. KClO4 is soluble in 100ml H2O to around 12 g @ 70 C and 22 g @100 C. K2SO4 is precipitated with saturated solutions to a small degree. The
nickel sulfate dissolves easily, the limiting material is the potassium perchlorate. Minimal water is best to ease precipitation of Ni(ClO4)2 later.
After all the perchlorate and sulfate are dissolved in the minimal amount of water, the solution is cooled and then ~double the equivalent volume of
90% isopropanol is added which precipitates the rest of the potassium sulfate. Remaining in solution is Ni(ClO4)2. Boiled down to minimal volume the
perchlorate crystallizes out on cooling and dried. I have been using this technique on the 100g scale in 1L vessels. I was very pleased that Ni(ClO4)2
can be prepared this way because it makes use of inexpensive KClO4 as the source of perchlorate and cheap isopropanol.
I am still learning about NAP. I'm using the "improved AGu" method of reduction from SM to derive AGu. It reacts well with this nickel perchlorate to
give a red reaction and the final NAP product in H2O at a 5 min boil. I have noticed that there seem to be different grades of NAP that have different
sensitivity and brissance. Apparently the darker red, the greater sensitivity and brissance. The lighter red (almost a reddish orange) detonates on
confinement and partially detonates unconfined but doesn't appear to detonate fully.
I have also noticed during the reaction, that a black solution forms in the reaction mixture after settling on cooling. The black mixture floats in
the middle with NAP layer both underneath and above it. I have been filtering the NAP early within around 20 minutes of cooling rather than waiting
the 4 hours suggested by the patent. I was afraid that this black solution would degrade the product. I'm not sure what this solution is, I think it
may be a further reaction with residual water in the solution. I will try this reaction in alcohols to see if it affects yield or prevents black
solution.
|
|
|
B(a)P
International Hazard
Posts: 1116
Registered: 29-9-2019
Member Is Offline
Mood: Festive
|
|
I have found the same thing both with the 'grades' of NAP and the black precipitate. In my experience the lower grade material you describe degrades
over time while the presumably more pure material appears to be stable when stored for at least a year in a sealed container.
|
|
|
Hey Buddy
Hazard to Others
Posts: 384
Registered: 3-11-2020
Location: Bushwhacker Country
Member Is Offline
|
|
Solvent interaction on NAP
I have just tried a few tests with alcohol solvents preparing NAP. The results were interesting so I will add here.
First two images are NAP patent procedure in terms of ratios and volume but I replaced H2O with 91% Isopropanol. The mixture begins as turquoise but
then changes to a red then at completion a brown black. The color change began at bp of isopropanol ~82 C.
The next four images are NAP reaction in commercial denatured alcohol solvent. The alcohol was brought to boil and there was no reaction. The volume
of alcohol (11ml) reduced over 5 minutes with no noticeable reaction. More alcohol was added to replace evaporated level. And as no reaction proceeded
10ml dH2O was added which immediately initiated a reaction which then proceeded through turning tan/brown, then reddish. At the end of the reaction
the (presumably) NAP separates and drops to bottom with a clear layer above. There is no black layer in this denatured alcohol method. When water is
used as the solvent there is a black color in solution and when isopropanol is used the black appears to mix with the product and create a dark
red/brown/black product.
Are there other solvents of interest? MeOH or Acetone?
I am also curious for other experimenters, how is the NAP coming out of solution for you? Is it dropping out immediately or forming slowly over the 4
hour sitting period? I ask this because all of my tests have resulted in immediate precipitation at conclusion of boiling. These pictures were all
taken following removal from heat, Im allowing them to stand 4 hours now.
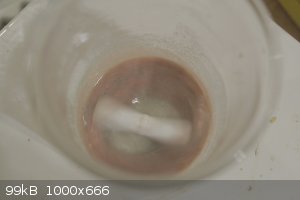     
[Edited on 13-4-2023 by Hey Buddy]
Update:
I just looked at these samples again after theyve been sitting at room temp for a few hours, the 50:50 denatured alcohol/added-H2O solvent test is
still free of black solution and the material is in a granular form with large grains visually. The isopropanol is a black/brown sludge.
[Edited on 13-4-2023 by Hey Buddy]
Update:
First image is comparison of the two materials on coffee filter paper. The denatured alcohol version is very granular and has homogenous color. I
thought it looked kind of less vibrant color red but in the second photo you can see its actually a shade lighter and deeper red tone than a
previously made sample shown in the vial. You can clearly see in the second picture the grainy consistency from denatured alcohol/water reaction.
normal consistency is powder.
 
[Edited on 13-4-2023 by Hey Buddy]
Is this color similar to what others are finding or is this color darker or lighter?
[Edited on 13-4-2023 by Hey Buddy]
The only other colors Ive seen for comparison are the videos from energetic heretic aka tetrazole lover. Its hard to tell what saturation of hue he is
getting. He has one picture that looks crazy red.
 
[Edited on 13-4-2023 by Hey Buddy]
UPDATE:
Its now the next morning and the samples have been dried. I tried burn tests of the samples. Again, I was surprised. The nice red color sample burns
unconfined by flame underneath. It turns black. It does not deflagrate or detonate. I'm a bit puzzled as to what is going on here. Direct hot ember
doesnt burn it either.
The less homogenous brown/black material from 91% isopropanol detonates ferociously in mg proportions without confinement. And also detonates
confined, which is logical.
I dont know what happened. I assumed for granted the red material was very pure and the black material was contaminated. I assumed the black material
would probably have performance issues. The experimental case was opposite. The black material DDT so powerfully. I think it is more brissant than
samples previously made by patent instruction. It is much more powerful than SADS or lead azide. I could feel the pressure energy in my skin from a
few feet away. And that is only from a small pinch. Incredible stuff.
[Edited on 13-4-2023 by Hey Buddy]
Just did a follow up test on the black material with 40 mg dropped loosely in a plastic vile, with 50% head space. The plastic vile was pressed into
dirt and a fuze was inserted. Detonation was quite powerful for such a small load. I could see dirt was fired into the air and the ~40mm hole made had
small shreds of plastic about the width of a fingernail clipping inside.
[Edited on 13-4-2023 by Hey Buddy]
Update:
This isopropanol NAP detonates ETN without problem and has very excellent flame DDT sensitivity. It can be detonated by direct visco contact without
any prime or thermite plug such as Zn/CuO (although Im sure a prime would increase reliability). I have had no failures to DDT in fuze tests and at
this time the original batch of black material is exhausted. Black NAP did not detonate a plastic vile of 15 g PETN. That test was a large plastic
vile of PETN with a small 8mm vile of black NAP, side-primed onto the PETN vile with black tape. The NAP was lightly tamped in the vile. That test
resulted in a failure to detonate the PETN. Im going to try to replicate isopropanol version and then scale up to 5g.
[Edited on 13-4-2023 by Hey Buddy]
|
|
|
Hey Buddy
Hazard to Others
Posts: 384
Registered: 3-11-2020
Location: Bushwhacker Country
Member Is Offline
|
|
I attempted to scale up the isopropanol method, using an equal ration of solvent but increased all portions from patent to 3x. I got a red paste this
time with less isopropanol boiling off at boil. Im going to first see if this batch high-orders then try to replicate a brown/black version again,
test a couple variations until I find consistency that is highest yielding and high power. Will follow up.
   
|
|
|
B(a)P
International Hazard
Posts: 1116
Registered: 29-9-2019
Member Is Offline
Mood: Festive
|
|
The colours that you are getting look ver close to what I have observed when doing this synthesis. I can also confirm that I get crystals coming out
of solution as soon as it starts to cool down. In fact I have noticed that yield seems to decrease if you leave it the suggested 4 hours. I generally
filter as soon as it has cooled to about room temperature. Looking forward to seeing how you go with the isopropanol.
|
|
|
Microtek
National Hazard
Posts: 828
Registered: 23-9-2002
Member Is Offline
Mood: No Mood
|
|
The black coloring is from decomposition of guanidine derivatives. It is one of the few drawbacks of this material that it decomposes if it is
dissolved in water. For this reason there is a balance to be found between adequate time to form the complex and too much time which will degrade your
product.
Hey Buddy: Please don't scale up production of primaries, even relatively safe ones such as this.
|
|
|
Hey Buddy
Hazard to Others
Posts: 384
Registered: 3-11-2020
Location: Bushwhacker Country
Member Is Offline
|
|
That's a sound warning. Unfortunately, it's too late for me.
--This material seems a little tricky in alcohol. I have now replicated the isopropanol brown/black sludge (I probably should not have described it
that way because it is more of a dark brown/red/violet/gray, not so much black). Anyways, I was able to recreate it and in this batch, it is certainly
degraded in performance. It's only partially DDTing. Not even damaging foil.
I also made an additional batch that was removed from boil (boiling in isopropanol is somewhat tricky because its boil is not obvious like water),
this reaction was removed before turning dark, it is mostly red but not as vibrant red as previous batches in past. It detonated at full power. This
is in contrast to the sample made earlier, which was very red, (made in denatured alcohol with H2O added after boiling). That sample is entirely
inert. It doesnt DDT at all. It simply decomposes to a sooty mass over flame.
So all in all:
-NAP is extremely powerful.
-It can be made in water or alcohol, likely other solvents.
-It is sensitive to time of boil, solvent type and molar ratios of reactants.
-It's possible to make inert NAP in both dark and light red forms.
-It's possible to make NAP that detonates secondary explosives in both dark and light red forms.
-It seems likely that a red product is desirable, and a darker product is decomposition. It is possible to make red product that is inert. It's not
recognized what is causing inertness in that case.
-***The black liquid typical of water solvent does not appear in alcohol solvents. This is the main difference between an H2O solvent and alcohol
reactions. It isn't known if there is even a benefit of using Alcohol instead of water. Experiments of equivalent molar reactions in water and alcohol
could reveal any benefit of using alcohol solvent, based on yield. It seems possible that alcohol solvent may increase yield but it needs further
investigation. In this experiment, producing inert material confused results a little bit, adding another layer of mystery.
Unknown:
-Do alcohols react with complex during boil?
-Exact molar ratios for maximum product.
I tried some isopropanol experimentation with near 2x excess Ni(ClO4)2 and it increased yield versus the stoichiometric ratio.
-How much boiling is appropriate with isopropanol? Alcohol boiling is a bit subtler than H2O so it has to be removed from stirring and visually
checked to identify when the boil starts. Appropriate time of boil may be less than 5 minutes for alcohols. The reaction can be removed from heat
during boil to choose the time of completion based on visual color change. Reaction continues mildly for minutes when removed from heat. A quench
might help stop reaction at desired color point exactly.
-It's unknown how long a darker product lasts. There are at least two reports of light red product remaining effective for a year and another over a
year, so it seems red product is desirable.
I'm very satisfied with this primary. It is much more powerful than lead azide and seems to be more flame responsive. Its preparation is very
efficient being a simply prepared complex. It would be interesting to see if bicarbs of AGu and Ni could be reacted with NH4ClO4 as the ClO4 ion
source. That would further simplify precursors.
I like it
I ordered a few kg of AGu bicarb.
  
[Edited on 14-4-2023 by Hey Buddy]
|
|
|
Hey Buddy
Hazard to Others
Posts: 384
Registered: 3-11-2020
Location: Bushwhacker Country
Member Is Offline
|
|
H2O vs IPA
Everything used in the test is mirrored. The difference between the two is only the solvent. dH2O is used as solvent in one reaction, 91% isopropanol
is used in the other.
Reagents used are:
1.38 g Aminoguanidine Bicarbonate .010 mol
1.85 g Ni(ClO4)2*6H2O .005 mol
15ml dH2O/91% IPA
The PID hot plates were preheated to 119 C. Stirring was set to 200 rpm and the respective reagents loaded into beakers along with solvents.
I refer to the reactions as "H2O" and "IPA" for shorthand.
1:49 The H2O begins to bubble and foam, IPA does not
2:14 H2O begins to change color, IPA remains
3:07 IPA begins to change color
3:37 Boiling is observed for H2O. 5 minute countdown started
7:14 Both H2O and IPA are very red in color
8:37 5 minute countdown complete, H2O and IPA removed from heat
24:25 H2O and IPA are poured out onto filters
Differences observed were
-IPA was noticeably hotter by handling the beaker than H2O through the 24 minutes of cooling.
-IPA appears to have precipitated a greater volume of material
-IPA has 2 colors of material, H2O has one very homogenous color
I will add the yields later and test their detonatability. Judging from this test, how consistent and uniform the H2O product is in appearance, H2O is
the way to go.
      
[Edited on 15-4-2023 by Hey Buddy]
UPDATE:
It is now the next day and both samples were dried for around 10 hours at 60 C. The H2O reaction yielded 1.185 g. The IPA reaction yielded 2.600 g.
Now comes the mystery part. The H2O reaction product does not detonate nor deflagrate unconfined, it is unreactive. The IPA reaction product
deflagrates unconfined similar to DDNP. This is in contrast to previous batches I've made which had nuanced preparation differences, both H2O and IPA
products in past have full immediate detonation unconfined.
I would assume the reagents are probably impure and introducing unknown contaminanation. These tests are done with in-house prepared reagents. I have
ordered some AGu and will repeat the test with that. I really dont want to buy Ni Perchlorate so hopefully that's not the contaminant. I will test if
Ni and AGu bicarbonate can be reacted with ammonium perchlorate as well. Which if successful, would be ideal. For my case, if nickel perchlorate can't
be produced without HClO4 it makes the production route less attractive to me than for instance lead azide. Especially with sabanajeff oxidation
route, and the volumes of data on lead azide. It's a very simple preparation route of only lead acetate, hydrazine sulfate and 50% nitric acid. It's
downside is toxicity and slightly under responsive to flame. I like the simplicity of SADS, but its under powered and reverts to silver and Mercury
Fulminate which is a bit sensitive. You can't both have cake and eat it too I suppose.
[Edited on 15-4-2023 by Hey Buddy]
|
|
|
Laboratory of Liptakov
International Hazard
Posts: 1339
Registered: 2-9-2014
Location: Technion Haifa
Member Is Offline
Mood: cool.gif
|
|
Interesting work. Hey Buddy, the Nickel aminoguanidine diperchlorate is hygroscopic ? A lot or a little? Or at all? Thanks...
Development of primarily - secondary substances CHP (2015) Lithex (2022) Brightelite (2023) Nitrocelite (2024)
|
|
|
Hey Buddy
Hazard to Others
Posts: 384
Registered: 3-11-2020
Location: Bushwhacker Country
Member Is Offline
|
|
I havent tested this but Ive had samples lay exposed to air for a weeks with no apparent moisture ingress or visible change.
|
|
|
Laboratory of Liptakov
International Hazard
Posts: 1339
Registered: 2-9-2014
Location: Technion Haifa
Member Is Offline
Mood: cool.gif
|
|
Thanks Buddy. Good message....
Development of primarily - secondary substances CHP (2015) Lithex (2022) Brightelite (2023) Nitrocelite (2024)
|
|
|
Herr Haber
International Hazard
Posts: 1236
Registered: 29-1-2016
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by Hey Buddy  |
I really dont want to buy Ni Perchlorate so hopefully that's not the contaminant.
[snip]
I will test if Ni and AGu bicarbonate can be reacted with ammonium perchlorate as well. Which if successful, would be ideal. For my case, if nickel
perchlorate can't be produced without HClO4 it makes the production route less attractive to me than for instance lead azide.
[Edited on 15-4-2023 by Hey Buddy] |
I'm pretty sure Ni perchlorate can be found at Onyxmet and Chemcraft if you change your mind.
I am fine with DLA too, but hydrazine / sodium azide is trickier to find than Ni(ClO4)2
The spirit of adventure was upon me. Having nitric acid and copper, I had only to learn what the words 'act upon' meant. - Ira Remsen
|
|
|
Microtek
National Hazard
Posts: 828
Registered: 23-9-2002
Member Is Offline
Mood: No Mood
|
|
LL: NAP is not hygroscopic at all, in my experience.
|
|
|
Hey Buddy
Hazard to Others
Posts: 384
Registered: 3-11-2020
Location: Bushwhacker Country
Member Is Offline
|
|
NAP advancement

I've been experimenting with methods of production of NAP. My interest has been to simplify its preparation and understand it's character further.
I am happy to say that this material has a lot of variation in its product and that isn't a detraction but a strength. It's possible to modify
sensitivity somewhat depending on purity, form etc.
I have found a route which prepares very high purity NAP in high yield. The nickel ion source is nickel carbonate and the perchlorate source is
ammonium perchlorate.
In my opinion, this route prepares the purest form of NAP other than using HClO4 directly. The added bonus is that with this route, each respective
material can be simply purchased and mixed in correct stoichiometry as needed.
Without capping agents or crystal modifications, this method produces entirely high purity, Blood-red, needle-shaped crystals. These crystals seem to
be more sensitive than other forms. This is opposed to NAP derived from Ni(ClO4)2*6H2O. which seems somewhat different in product. This also differs
entirely from Ni(ClO4)2 produced from NH4ClO4 or KClO4.
Previous preparations, such as Ni perchlorate via NiSO4 have considerable surface contamination by sulfate ion which appears to modify crystal to be
less mechanically sensitive and spheroidal, which may be desireable or not.
Other preparations of Ni perchlorate as a precursor have been found to be entirely unnecessary and in fact, less efficient and lower yielding than the
method of simply mixing correct ratios of NiCO3, NH4ClO4 and AGuHCO3, followed by heating.
NiCO3 can be bought from ceramic supply stores world wide. It is used as a glaze component in kiln fired pottery. Here in the USA, I have found it
around $20/lb and it is widely available from many suppliers.
I am still experimenting with crystal modification but was very satisfied with this method of production. In my opinion with this method, NAP becomes
the most desirable primary explosive as it yields well, is high performance, and its preparation is simpler than even SADS once this method is used.
With ammonium perchlorate production being possible at home, the various routes to Aminoguanidine and carbonates available as ceramic glaze
components, this is the most home-lab practical primary, in my opinion.
In testing, I have found NAP to be the swiss army knife of primary explosives, being very capable. All of the excitment i've read on SM about this
complex is justified.
I believe it could easily displace legacy primaries and will no doubt, in amateur production, displace current primaries, only a matter of time.
I've found nothing comparable.
|
|
|
B(a)P
International Hazard
Posts: 1116
Registered: 29-9-2019
Member Is Offline
Mood: Festive
|
|
Well done Hey Buddy! Agreed with your sentiments on the ease of production and performance. Looking forward to seeing your nickel carbonate route when
you have it optimised.
The best yield I got with the nickel perchlorate / aminoguanidine bicarbonate route was about 50%, very curious to see your yields via nickel
carbonate.
|
|
|
MineMan
National Hazard
Posts: 998
Registered: 29-3-2015
Member Is Offline
Mood: No Mood
|
|
Those are giant crystals! Great work Hey buddy
|
|
|
Diachrynic
Hazard to Others
Posts: 219
Registered: 23-9-2017
Location: western spiral arm of the galaxy
Member Is Offline
Mood: zenosyne
|
|
I'm curious for a proper writeup, since if you have NiCO3, you can make the acetate from it directly and use the preparation via that. It
will be interesting what gives a higher yield (and especially if some reagent has to be in excess).
These microscope images are old, but still fairly pretty:
   
The corresponding nitrate has a totally different crystal morphology, and the sulfate on account of being quite insoluble in water doesn't form good
crystals at all. It would be nice to find a salt that is as pretty and crystalline as the perchlorate, but without being energetic - it would make for
a good beginner friendly experiment in coordination chemistry.
we apologize for the inconvenience
|
|
|
Microtek
National Hazard
Posts: 828
Registered: 23-9-2002
Member Is Offline
Mood: No Mood
|
|
You can avoid the formation of long needle crystals (which, while pretty, are less safe and more difficult to process) by maintaining stirring
throughout the precipitation phase. It's probably possible to tweak the morphology by adjusting the RPM of the stirrer.
|
|
|
Hey Buddy
Hazard to Others
Posts: 384
Registered: 3-11-2020
Location: Bushwhacker Country
Member Is Offline
|
|
| Quote: Originally posted by Microtek | | You can avoid the formation of long needle crystals (which, while pretty, are less safe and more difficult to process) by maintaining stirring
throughout the precipitation phase. It's probably possible to tweak the morphology by adjusting the RPM of the stirrer. |
I saw that you mentioned this earlier in the thread and can confirm. RPM adjustment on cooling does change crystal size. At 200 rpm Im finding about
the same size as with IPA.
I've tried some different crystal modifications:
-Propylene Glycol solvent: reacts with complex, red/brown syrup results.
-CMC: prevents complexation
-Dextrin: prevents complexation
-CMC: added after complexation, diminishes yield, impractical, short blocky crystals
-Dextrin: added after complexation, diminishes yield, long needle crystals are still formed
-Isopropanol solvent 90%: changes time of boil of reaction, lower boiling point, longer reaction time, complex crystallizes as a paste/powder with a
lighter salmon color, apparent reduction in mechanical sensitivity
IPA yields are about +40% over H2O solvent. Beginning with a 1 g AGu reaction, H2O results .97 g, IPA results in 1.38 g
There is always green nickel remaining in filter after reacting stoichiometrically in H2O, IPA appears to leave less Ni in filter. Ni may benefit from
reduction to less than stoichiometric in order to reduce unreacted Ni in product.
[Edited on 11-5-2023 by Hey Buddy]
|
|
|
| Pages:
1
2
3
4
5 |








