2,4-Dinitroanisole
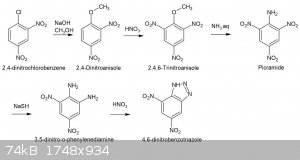
The ultimate target of this synthesis is 2,4,6-trinitroaniline (picramide) and thence to 4,6-dinitrobenzotriazole via 3,5-dinitro-o-phenylenediamine.
Picramide can be prepared by numerous routes 1),4) but is most conveniently prepared by treating trinitroanisole with ammonia solution 2). The
trinitroanisole being prepared by the nitration of 2,4-dinitroanisole 3) but the sequence begins with the methoxylation of 2,4-dinitrochlorobenzene
and it is this part of the preparation that I wish to report here:
The preparation of 2,4-dinitrochlorobenzene has been discussed elsewhere on this forum and since I have a significant amount of this material, I will
use it as the starting point. The analogous bromo- compound may be used and its preparation is illustrated in a Youtube video 5). I am currently
working on a third, 100g scale preparation with photographs for the pre-publication section.
The best detailed description of the synthesis I could find was in Urbanski’s book Vol.1 4), however, the details are for an industrial scale
process start with 600kg of 2,4-dinitrochlorobenzene! The procedure presented in Urbanski’s book calls for 800kg of methanol, 600kg of
2,4-dinitrochlorobenzene and 196kg of caustic soda! The method was scaled to 10g of dinitrochlorobenzene and the other components reduced pro-rata. In
my first attempt I scaled this down to just 10g and encountered a few problems, the reaction mass is a thick paste that can’t be stirred, it’s
more of a kneading process; the reaction time of 10-11 hours seem excessive in the light of my experience and the product is deep yellow suggesting
the presence of much sodium dinitrophenolate. It is assumed that from an industrial perspective the small amount of methanol used may be an advantage.
In my second attempt I decided to use homogeneous reaction conditions with all ingredients in solution. This I hoped would create better contact
conditions between reagents and simplify the work-up to simply filtering of the sodium chloride formed and diluting the reaction mixture to recover
the dinitroanisole. To my delight the synthesis proved to be unbelievably facile. The initial reaction is vigorous, almost instantaneous, it is not
necessary to filter of the salt as this remains in solution when the reaction mixture is diluted with water and there is almost no dinitrophenol
formed during this initial reaction according to TLC. The yield excellent >94% and a little more, less pure, material was recovered from the
filtrate after recovering the methanol by distillation.
Experiment 1
10.008g of finely powdered 2,4-dinitrochlorobenzene were added to 3.300g of sodium hydroxide dissolved into 19ml of warm (40°C) methanol in a
tall-form beaker suspended in a water both at 40-45°C. The sodium hydroxide did not dissolve completely due to the presence of sodium carbonate. The
powdered benzene derivative was added in seven small portions of about 1.5g and the mixture manually stirred as it rapidly became a thick paste. Upon
each addition a deep red colour appeared momentarily and then faded, being replaced by a mustard-yellow sandy precipitate. The addition of the solid
dinitrochlorobenzene is accompanied by a mild exothermic reaction and if added too quickly local boiling and sputtering occurs but the temperature is
easily maintained at 45-50°C with care. When the addition was complete the mixture was stirred as best as possible but by this point the mixture is a
semi-solid mass and mixing is difficult. The mixture was warmed in a water bath to 50-55°C for three hours with periodic manual stirring.
After 3 hours heating the mixture was diluted with an equal volume of water, chilled and filtered in a small Buchner funnel. The deep yellow cake was
washed several times with water after isolation of the initial filtrate. The washings were deep yellow and stain the hands a clear sign that
nitrophenols are present. The cake was slurried in a small beaker with fresh water and filter again, washed with cold water and dried to give 9.365g
of bright yellow solid. TLC (8:1 Pet. ether:ethyl acetate) showed three spots, the main spot of dinitroanisole at 0.12, a faint yellow spot at 0.08
and a faint crescent at about 0.4 of unreacted starting material.
An experiment revealed that the compound can be recrystallised from 95% ethanol using 11ml per gram and gives about 83% recovery. The raw material was
recrystallised from 80ml of rectified spirit, treated with a little charcoal and filtered hot. The product crystallised in pale yellow needles that
gave only a single spot on a TLC but the recovery was only 4.360g suggesting that the charcoal treatment was excessive.
Experiment 2
5.28g of (approximately 50% molar % excess) sodium hydroxide was dissolved in 25ml of warm methanol (40-45°C); it did not dissolve completely
probably due to the presence of some sodium carbonate but this is not a problem. 20.061g of 2,4-dinitrochlorobenzene were dissolved in 50ml of warm
methanol and then the sodium hydroxide solution added too it slowly. A vigorous, mildly exothermic, reaction occurs with a transient red colour
rapidly fading and a dense pale ppt forming. If the addition is too rapid brief boiling may occur but this does not appear to affect the outcome,
mechanical stirring also helps to avoid localised boiling. Once addition was complete the suspension as maintained at 50°C for about 20 minutes and
then allowed to cool, during which time the suspended solid (salt) settled to the bottom. A TLC taken at about 10 minutes revealed only dinitroanisole
and a trace of the yellow compound.
By the time the reaction mixture had reached room temperature it had almost solidified to a very pale-yellow mass. This was stirred up with an equal
volume of cold water and then chilled in the fridge for several hours before being filtered. The solids filter easily as the ppt is crystalline. The
cake was washed on the filter and then transferred to a beaker with about 150ml of cold water and filtered again. The cake was washed and drained as
much as possible and then dried at 45-50°C to constant weight to give 18.586g of very pale straw coloured raw 2,4-dinitroanisole (94.7% of theory)
but it is homogeneous by TLC.
The initial filtrate was distilled to recover about 50ml of aqueous methanol and when the cold deposited a further 0.410g of deep yellow fibrous
solid. TLC reveals two major components, the dinitroanisole and the yellow compound with Rf value of 0.08; presumably 2,4-dinitrophenol.
Discussion
In the Japanese process produced in Urbanski’s book the reaction period is roughly 10-11 hours and in my experimental method based on this procedure
there was clearly a little unreacted 2,4-dinitrochlorobenzene as revealed by TLC in the raw product after 3 hours. The problem seems to be caused by
the difficulty of getting good contact between the reagents in an environment that is never homogeneous. This is compounded by the difficulty in
stirring the thick paste.
By contrast, when starting with warm, homogeneous solutions in methanol the reaction is almost instantaneous. Other advantages of this procedure are
the smaller amount of dinitrophenol produced, ease of stirring and simplicity of the final work up.
Conclusion
The methoxylation of 2,4-dinitrochlorobenzene to 2,4-dinitroanisole is best carried out in homogeneous solutions, the reaction is rapid, facile and
the product pure enough for subsequent nitration without further purification.
1) Mee A. J.; Richter’s chemistry of carbon compounds 3rd Ed.; vol 3, p103 and references there in.
2) Salkowski H.; 1871; Berichte der Deutschen Chemischen Gesellschaft v4, p873
3) Urbanski T.; 1964; Chemistry and technology of Explosives vol. 1, p541
4) Urbanski T.; 1964; Chemistry and technology of Explosives vol. 1, p558
5) https://www.youtube.com/watch?v=qTYpHY1Nka0
|









