| Pages:
1
2 |
clearly_not_atara
International Hazard

Posts: 2722
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
Quote: Originally posted by Pumukli  |
Interesting what you do with these triazoles and unveiling a possible otc source would be a nice teaser too. :-) |
I think you could obtain o-phenylenediamine from an Ullmann-Goldberg reaction between o-dihalobenzene (or equivalent) and urea to
o-phenyleneurea followed by hydrolysis. If you really want benzotriazole, this should work fine, since phenylenediamine + nitrous acid works pretty
reliably. See e.g.:
http://www.sciencedirect.com/science/article/pii/S0040403904...
[Edited on 4-11-2015 by clearly_not_atara]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
This patent lists several nitration schemes for benzotriazole, seems that depending on the conditions of nitration and/or protection group addition,
several different isomers are possible in good yield. The temperatures for nitration seem very harsh though, anyone has experience with nitration of
benzotriazoles?
[Edited on 11-12-2015 by nitro-genes]
Attachment: US20040234958 Benzotriazole nitration.pdf (2.4MB)
This file has been downloaded 530 times
|
|
|
Boffis
International Hazard
Posts: 1843
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
@ nitro-genes. Thank you for this patent. What an amazing collection of interesting triazole chemistry! I have to try some of these preparations. I
haven't tried nitrating benzotriazole yet but I acquired some 5-methylbenzotriazole which I hope will nitrate more readily. I also tried the
preparation of 5-aminotriazole from 2-nitro-1,4-phenylenediamine but so far I haven't manage to isolate the initial 1,2,4-triaminobenzene. The problem
is that it is very sensitive to oxidation so I tried to keep the solution acid with HCl to recover it as the hydrochloride which I hoped would be more
stable. I will try again using the protocol from Org Synth for 1,2-phenylenediamine under alkaline conditions.
I will also try to make 4-substituted o-phenylene diamine derivatives from 4-substituted anilines and p-aminobenzoic acid. 2,4-dinitroaniline is
selectively reduced to 4-nitro-1,2-diaminobenzene by sulphide ions (Org Synth again) and coupling diazotized sulphanilic acid with p-toluidine,
4-nitroaniline and p-chloroaniline followed by reduction with dithionite will (hopefully) give me the 4-methyl, 4-nitro and
4-chloro-o-phenylenediamine and hence the appropriate triazole. Does any one out there have any experience of rearranging azoamino to amino-azo
compounds? It used to be a standard Uni. 1st year prep but they always used aniline so the product was the 4-aminoazophenyl derivative were as I am
trying to get the 2-amino-azo derivative by blocking the 4 position; will it work?
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Interesting stuff you said there regarding the azo-amino rearrangment.  The
interaction between different substitutions on the ring and the behaviour of the corresponding nitro/nitroso/nitr(so)amine/(di)azo(xy) groups under
different conditions is amazingly complex. the more you read about it, the more complicated it seems to become. My guess would be that the p-amino
group of 1,2,4-triaminobenzene is likely to lead to a lot of side reactions, since only in the case of an o-diamine the intramolecular rearangement
after diazotization to the triazole would be favored. How stable is triazole to NaHS reduction and basic conditions, guess there is a reason you want
to go via the triamino? The
interaction between different substitutions on the ring and the behaviour of the corresponding nitro/nitroso/nitr(so)amine/(di)azo(xy) groups under
different conditions is amazingly complex. the more you read about it, the more complicated it seems to become. My guess would be that the p-amino
group of 1,2,4-triaminobenzene is likely to lead to a lot of side reactions, since only in the case of an o-diamine the intramolecular rearangement
after diazotization to the triazole would be favored. How stable is triazole to NaHS reduction and basic conditions, guess there is a reason you want
to go via the triamino?
Regarding the 4-substituted o-phenylenediamines, would benzimidazolone be a good starting point? n-methyl benzimidazolone can be found OTC. The two
para positions relative to the aminogroups would be most activated. Hell, it might even nitrate all the way to the tetra nitro and produce an
tetranitrotriazole. 
[Edited on 16-12-2015 by nitro-genes]
|
|
|
Boffis
International Hazard
Posts: 1843
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
This thread has been a bit quiet recently so I thought I had better revitalize it a bit. I have been working on preparing substituted o-diamines and
it has proved a good deal trickier than expected. The main issue is the sensitivity of these compound to oxidation. My current target intermediate is
3,4-diamino-N,N-dimethylaniline. This compound should convert fairly easily into a triazole but it is also of interest as one of the most sensitive
reagents for selenite ions, in acid conditions it give a red colouration with selenite but not with selenate, tellurite, sulphite etc.
My proposed method to prepare it starts with dimethylaniline because I have some and its 4 position is very reactive. The route is:
1) Dimethylaniline + NaNO2 + excess HCl = 4-nitrosodimethylaniline hydrochloride in almost quantitative yield
2) 4-nitrosodimethylaniline hydrochloride in HCl sol + tin = 4-amino-dimethylaniline Hydrochloride (no isolated)
3) neutralise with NaOH and extract with ether
4) remove the ether and add glacial acetic acid and acetic anhydride = 4-dimethyamino acetanilide
5) Nitration of 4-dimethylamino acetanilide with ?? = 2-nitro-4-dimethylamino-acetanilide
6) reduction of 2-nitro-4-dimethylamino acetanilide with tin + HCl = o-diamine hydrochloride
7) Liberate free amine with NaOH, extract with ether and rapidly extract back into HCl and evaporate down to crystallise the desired compound as the
hydrochloride or if the triazole is the desired product then treat directly with sodium nitrite.
I have made several attempts using 5g of dimethylaniline at a time. The nitrosation and reduction work well but the isolation of the amine is tricky
because the tin salts produced a precipitate when the solution is made alkaline. Fortunately this precipitate largely remain in the aqueous phase
during the ether extraction. The larger problem is the speed at which the free amine oxidizes and darkens even in ether solution. I have found that
after the the recovery of most of the ether the remainder can be treated with 1ml of acetic anhydride (an excess) and 2.5ml of glacial acetic acid per
gram of crude amine this makes drying the ether solution unnecessary. In another experiment I used an even larger excess of acetic anhydride and no
acetic acid, this worked but the final acetanilide derivative is more discoloured.
When the acetylating mixture is has been heated to reflux for 20 minutes it was cooled and poured into cold water but no precipitate forms. I have
found that the acetanilide is sufficiently basic to dissolve in dilute acetic acid and so the solution must be made slightly basic with NaOH solution
and then the acetanilide filter off. It may be recrystallised by dissolving in 2.5ml of isopropanol per gram with working and diluting with 7ml of
water per gram and chilling in the fridge. Slowly thin, very pale lilac, pearly scale crystallise out in good yield. If the compound is excessively
discoloured (as happens when the synthesis to this point has taken more than a few hours) charcoal may be added after dilution and the solution
reheated before filtering.
I will post the experiment details shortly but does any one have any ideas about the reduction of the nitroso-compound that will not deposit
hydroxides when the solution is neutralised? If this could be achieved it would remove the need to use ether extraction, the precipitated free amine
could simply be filtered off, suck as dry as possible and immediated acetylated with excess acetic anhydride.
The next stage is nitration, I intend to try using the procedure in Vogel for the nitration of acetanilide unless anyone has any better ideas. I
haven't gotten to thinking about the reduction to a diamine yet but but I may try the H2S or alkali sulphide as in the Org Synth reduction of
2,4-dinitroaniline to 4-nitro-1,2-phenylenediamine.
I haven't tried to prepare the dimethylamino-1,2-phenylenediamine from the mono-amine by coupling with diazotised sulphanilic acid and then reducing
with thiourea dioxide or Na dithionite yet because of the instability of the amine, I think it will act as a reducing agent and simply convert the
diazonium compound to benzene sulphonic acid.
Incidently I tried using the 2-nitro-1,4-phenylene diamine that I mentioned earlier but the resulting triamine or whatever is formed is insanely
unstable and I alway end up with a brown gunk at the end of the reduction but this may be due to the highly oxidized nature of the old starting
material.
|
|
|
clearly_not_atara
International Hazard
Posts: 2722
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
I'm pretty sure step 5 is wrong; it produces 3-nitro-4-dimethylaminoacetanilide. Consider instead perhaps the nitration of 4-fluoroacetanilide
followed by reaction with dimethylamine?
https://en.wikipedia.org/wiki/Carbendazim and other benzimidazole fungicides may be ideal starting materials for o-phenylenediamine synthesis.
Benzimidazoles can also be used as intermediates rather than precursors: some N-phenylamidines undergo chlorination leading to nitrene insertion and
the formation of benzimidazole. This synthesis of 2-phenylimidazole:
http://140.123.79.90/~seekwei/thesis-reference/41-1932_ftp.pdf
suggests that benzimidazoles may be easily made from aniline and phenyl cyanide. Hydrolysis produces o-diaminobenzene, no nitrations required
[Edited on 5-12-2016 by clearly_not_atara]
|
|
|
Boffis
International Hazard
Posts: 1843
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Hi clearly_n_a, on reflection you may be correct but I an going to try it anyway and see what I get! Incidently may belief in this approach isn't
random. Nitrosation of dimethylaniline yields the 4-nitroso compound but if this position is occupied no reaction take place so while the
dimethylamino group is strong p activating it doesn't seem to affect the o positions to the same extent for whatever reason. Also nitration of
4-acetylaminophenyl acetate yields the 3-nitro compound.
One point I should make is that I am trying to prepare substituted o-phenylenediamines. I already have a lot of the simple o-phenylenediamine already
but its difficult to substitute other groups into it because its so sensitive to oxidation (though much more stable than the p isomer). The simplest
one to prepare is 4-nitro-o-phenylenediamine from 2,4-dinitroaniline via sulphide reduction (Org Synth procedure)
I don't have any 4-fluoraniline tough I have plenty of 4-nitroaniline and sodium tetrafluoroborate so I could probably make some; do you think that
4-chloro or 4-bromoaniline would work? I would have expected that that the bromo group would be easier to replace? I also have some
2-nitro-4-chloroaniline but replacing the chloro group is very difficult.
Your suggestion regarding reacting aniline with phenylcyanide is interesting and worth a try using aminodimethylaniline and phenylcyanide, I'll look
into this and report back.
|
|
|
Boffis
International Hazard
Posts: 1843
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
This thread has been dormant for a long time but I have not. I have continued to read up on this field and I have found many interesting paper on
triazole chemistry, mostly in German, and it appears that unlike other benzoazole (eg benzimidazole) benzotriazole nitrates easily in the 4-position
and this can be reduced to the 4- amino compound from which many more compounds are available. I have translated some of the more important papers and
will post these over the coming weeks along as appropriate. I have also found that most of the chemistry speculated on above has in fact already been
done, so for instance picrylamine can be selectively reduced to 3,5-dinitro-o-phenylenedimine and then treated with nitrous acid to give
4,6-dinitro-benzotriazole. This was in fact one of the first triazoles to be prepared and by Griess, the founder of diazo chemistry.
|
|
|
Boffis
International Hazard
Posts: 1843
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Nitration of Benzotriazole
I have finally gotten around to experimenting with the nitration of benzotriazole, its only taken 5 years. This is still work in progress and I need
to prepare the 5-nitro isomer by another method, as described above, to check the identity of the two main products. I then plan to reduce it to the
amine and investigate the interesting array of derivatives described in the references at the end and others not included yet. I also intend to
investigate the possibility of further nitration to 5,7-dinitrobenzotriazole which can also be prepared from picric acid via picramide
(2,4,6-trinitroaniline) and 3,5-(4,6)-dinitro-o-phenylenediamine 4) as discussed in earlier posts. I then intend to post the final write-up in the
Pre-publication section and this version will contain translations of the more important German references.
Nitration of Benzotriazole
Boffis January 2022
According to the work of Fries et al1) benzotriazole is nitrated exclusive in the 4 position e.g. the position adjacent to the fused triazole ring.
The 5 isomer is easily prepared by indirect means from 2,4-dinitroaniline via the selective hydrosulphide reduction to 4-nitro-1,2-phenylene-diamine2)
and in better detail3).
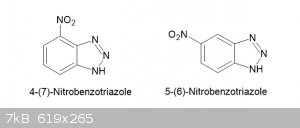
The method described below was developed from the method of Fries et al (p229) and worked admirably but TLC of the product revealed that it is a
mixture of 3 compounds. One of them, an orange-coloured material occurs in only trace amounts but the other two are significant components, the
purification process suggests a ratio of about 3:1. The more abundant component forms tiny, blocky, very pale-yellow crystals and is presumed to be
the 4-nitrobenzotriazole while the minor component forms slightly dark yellow fibrous aggregates and fits Zincke’s description of the
5-nitrobenzotriazole but could be in part at least a dinitro compound. The two compounds are easily separated due to their large difference in
solubility, particularly in acetone and butanone.
Experimental
Preparation scale nitration of commercial benzotriazole
72.12g of crude commercial benzotriazole granules were added fairly quickly to 205ml of technical grade concentrated sulphuric acid with gentle
stirring. The benzotriazole dissolves quickly and the temperature rises to about 60°C. The light brown solution was cooled to about 5°C and then
placed in an iced water bath on a stirrer plate (note 1). 44ml of concentrated (69%) nitric acid (approximately 10% excess) were added slowly from a
dropping funnel at such a rate that the temperature did not rise above 30°C.
 
Figure 1; crude benzotriazole (left) and benzotriazole dissolving in sulphuric acid (right).
If the temperature starts to rise too quickly place the beaker in a freezer for 20-30 minutes until the temperature has dropped to about 0°C and then
continue the addition of nitric acid. When the addition was complete the beaker was allowed to stand at room temperature for 20 minutes and then
warmed to 55-60°C slowly over 30 minutes to complete the reaction.
The clear amber coloured solution was allowed to cool to 15°C and poured over 100g of ice and the beaker rinsed with about 25ml of cold water. The
hot, pale yellow, suspension was allowed to cool to room temperature (about 8°C) and 200ml of 50% sodium hydroxide added slowly over about 5 minutes
(note 2). The suspension become exceedingly hot and was allowed to cool again to room temperature overnight (to about 5°C).
 
Figure 2; fully dissolved benzotriazole solution in H2SO4 (left) and after addition of the nitric acid.
 
Figure 3; the reaction mixture after heating for 30 minutes, ready to drowned and the drowned reaction mixture.
The suspension was filter using a large (12.5cm) Buchner funnel, washed with a little water and then pressed down and sucked as dry as possible, any
cracks in the cake healed with a spatula. The pale straw-yellow cake was dispersed into 350ml of cold water and then stirred for 15 minutes until a
thin cream-like suspension had formed. The suspension was filtered again and sucked as dry as possible and then air dried at 35-40°C to constant
weight on two 200mm watch-glasses, this took nearly 2 days to achieve. The yield was 80.81g or 81.3% of theory (note 3). A TLC revealed that the raw
product is a mixture of three compounds. A faint orange line visible without UV is only a trace impurity but the other two are both significant
components.
 
Figure 4; filtering off the nitration product, note the clear filtrate (left) and the still-moist cake of crude product.
The crude nitrobenzotriazole was purified by the following method: 38.5g of crude product were boiled in a 500ml conical flask with 195ml of acetone
(5ml per g) briefly and then the suspension allowed to cool to room temperature. The pale pinkish yellow crude 4-nitrobenzotriazole was filtered off
washed with a little (25ml) acetone and dried to give 25.347g. The filtrate, about 220ml, was distilled to recover 110ml of acetone and cooled to room
temperature overnight. The gritty, pale-yellow crystals of fairly pure 4-nitrobenzotriazole were filtered off and dried to give a further 3.318g of
4-nitrobenzotriazole. The remaining filtrate was distilled to give about 50ml of orange liquid that was poured into a glass bowl and allowed to cool
and evaporate. When evaporation was complete and the smell of acetone gone a pale orange granular material filled the bottom of the bowl while around
the edge was a brighter pinkish orange efflorescent crust. The granular material was carefully removed leaving the efflorescent rim adhering to the
edge of the bowl. The granular material is believed to be mainly 5-nitrobenzotriazole and weighed 8.436g. The efflorescent rim was carefully removed
and weighed 0.601g appears to be a mixture of the 5-nitro isomer and a delicately fibrous orange compound. The missing 0.8g is the result of
mechanical and handling losses that are difficult to avoid with hot acetone solutions because of their low viscosity and surface tension and the need
to work quickly.
The crude 4-nitrobenzotriazole can be recrystallised from boiling acetone or butanone (20ml per gram) to give very pale-yellow blocky crystals in 55%
yield without working up the filtrate. Other solvents invariably give a woolly fibrous product that is much less convenient although the single-crop
yield may be better e.g. 18ml per gram of 95% ethanol gives a single-crop yield of about 76% but of fine, matted fibres. Work is still in progress on
the two minor products but methanol (6ml per gram) appears to be the best solvent for recrystallising the granular 5-nitro isomer, the yield is about
90% but it still appears to be a mixture of scaly rosettes (the major product) and tiny fibrous pompoms that resemble the 4-nitro compound when it is
recrystallised from ethanol.
Note 1; With small scale experiments using 20g of benzotriazole ice-water is adequate but for this larger scale preparation it would be better to use
an ice-salt bath and probably cool the starting solution to -5 or below first.
Note 2; nitrobenzotriazole is fairly soluble even in dilute sulphuric acid, the amount of sodium hydroxide neutralises most of the sulphuric acid to
sodium hydrogen sulphate greatly reducing the solubility of the base. Further neutralisation to neutral sodium sulphate is best avoided since large
amounts of it crystallise out complicating the work-up and reducing yield through mechanical losses as I have discovered in earlier experiments.
Note 3; Attempts to recover more material from the original sodium hydrogen sulphate filtrate proved worthless, less than 0.3g were recovered by
further neutralisation and decanting from the sodium sulphate crystals through a filter. The mixture of nitrobenzotriazole and sodium sulphate hydrate
being washed with a little water and the residue dried.
1) K. Fries, H. Güterbock and H. Kühn; Annalen der Chem.; Investigations into the series of benzotriazole and N-methyl-benzotriazole; p213-240
(1934)
2) A. W. Hofmann; The action of nitrous acid on nitrophenylenediamine; Annalen der Chem; v115, p249, (1860)
3) T. Zincke; On Chloroketones and quinones of heterocyclic compounds and their derivatives; Annalen der Chem., v311, is3, pp276-329 (1900)
4) Norton & Elliot; Dinitro-1,2-diaminobenzene; Berichte v11, p327 (1878)
|
|
|
Boffis
International Hazard
Posts: 1843
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
I am still investigating the nitration of benzotriazole and now also benzimidazole but I am having a problem with TLC. These nitrobenzo-heterocyclic
are often very polar and simply sit at the origin and the spots hardly move at all. I am currently using a mixture of petroleum ether and ethyl
acetate (6:1). Does anyone have experience of such polar compounds and can suggest a better solvent for them?
[Edited on 4-4-2022 by Boffis]
|
|
|
AvBaeyer
National Hazard
Posts: 645
Registered: 25-2-2014
Location: CA
Member Is Offline
Mood: No Mood
|
|
If I am not sure where to start in terms of solvent polarity, I make the first run with 1:1 EtOAc:hexane then modify the solvent system from that
initial observation. If pure ethyl acetate is not working, then I move to chloroform and increase solvent polarity with methanol a few per cent at a
time. For really polar compounds I use ethyl acetate-butanol-acetic acid-water mixtures. Sometimes chloroform-hexane mixtures work well also with
polar compounds like phenols.
AvB
|
|
|
Boffis
International Hazard
Posts: 1843
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
@ AvBaeyer; many thanks for your helpful comments. I have now played around and found that chloroform-methanol seems to work well and I have settled
for a 5:1 ratio. Interestingly this mixture has resolved the original nitration product and, more clearly, the final residue from recrystallisation
into 5 spots!
I am very please with my final 4-nitroproduct which gives only a single spot. I am now working on recovering the other product which I presume to be
mainly the 5 isomer.
I forgot to mention that the TLC I used in the original prep were developed with pure ethyl acetate but this causes streaking so the upper streak can
only be resolved into 2 bands. The lower streak from the origin to about 0.4 is the yellow impurity. With the chloroform methanol solvent it resolves
into a single fairly sharp band at about .15 so I'm pleased .
|
|
|
mayko
International Hazard
Posts: 1218
Registered: 17-1-2013
Location: Carrboro, NC
Member Is Offline
Mood: anomalous (Euclid class)
|
|
I did the limonene -> carvone synthesis a while back and I saved samples of the intermediates. I was looking for ideas for what to do with them and
ran across this paper where they attach various nitrosochlorides to various nitrogen heterocycles, including benzotriazole. Not sure where that gets
you, but there it is.
Attachment: Reactions of 3-carene limonene and a-pinene nitrosochlorides with imidazole benzotriazole and indole.pdf (238kB)
This file has been downloaded 131 times
al-khemie is not a terrorist organization
"Chemicals, chemicals... I need chemicals!" - George Hayduke
"Wubbalubba dub-dub!" - Rick Sanchez
|
|
|
| Pages:
1
2 |










