| Pages:
1
2
3
4 |
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Quote: Originally posted by Hennig Brand  | No, it did not get the proper color before the addition of the sodium azide. It did go a bit orange but not as much as the description in the
synthesis said it should. I forgot to mention that.
I used the proportions and procedure from the Mr. Anonymous synthesis posted at the start of this thread.
I have made a little picric acid directly from phenol, we will see if that makes a difference. I will try and do a little testing tomorrow.
|
It sounds like a materials or possibly a pH issue. The azo-clathrate is a basic lead salt so alkalinity is critically definitive here, and if it isn't
just right ....well uh oh, it's right  or it isn't right or it isn't right  .....you know how that one goes, if it isn't right then you get no joy on the result. .....you know how that one goes, if it isn't right then you get no joy on the result.
[Edited on 14-8-2011 by Rosco Bodine]
|
|
|
Hennig Brand
International Hazard

Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
I think I may have solved any possible materials problems with a fresh batch of picric acid from phenol. The problem is definately before the sodium
azide addition I would say, probably ruling out any problem with the sodium azide.
As for the pH issue, you mentioned something to me in the picric acid thread last year about titrating a solution of NaOH for greater accuracy. I can
make up a solution of sodium hydroxide and titrate it to find its exact concentration. Titrating against hardware store muriatic acid should get me
pretty close, don' t you think? This is what I will do if it comes to that.
I am still pretty sure my picric acid is heavily contaminated. I think I may have oxidized some of the ASA during sulfonation.
[Edited on 14-8-2011 by Hennig Brand]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Yeah it is a lot more accurate weighing a larger quantity to make a stock solution
as a standard, a manually calibrated solution which can be dispensed more accurately by volume to provide the needed equivalent, than is the
alternative manipulation of weighing out a small amount of especially an air and moisture sensitive material like NaOH which may have unknown
composition right from the bottle. Actually it was no coincidence that I linked to some useful laboratory solution tables from Merck ....sort of a
little hint there The weight of NaOH can be hit close enough by direct
measuring done quickly with pure reagent and accurate scales .....but really using a standard solution is the proper way of doing measuring like this
so you know for sure what equivalent you are measuring out while dispensing material for an experiment to know it is in fact an accurate amount. If
you do things carefully for much work pH indicators aren't needed
except for confirmation analyses on the stock solutions. Food grade sodium bicarbonate is a better choice really for dry measure, especially on a
humid day.
I often will substitute sodium bicarbonate where that is no problem and better accuracy is needed and that should work fine here, at least to the
point of neutralization of the picric acid, and then the extra alkalinity can be gotten with NaOH stock solution or "dry" measure of NaOH to reduce
the error. I have used all three methods pretty much interchangeably without much thought about it.
A very good and accurate standard is fresh 1280 battery electrolyte, which generally from the manufacturer is absolutely pure and dead on accurate,
and easy enough to double check against a bicarb titration. This is close
enough for anything you are likely to ever do in a lab.
|
|
|
krazypunk50
Harmless
Posts: 16
Registered: 10-7-2010
Member Is Offline
Mood: Audacious
|
|
Should the synthesis of the azo-clathrate be done under reflux as to not lose any water? or is that unnecessary?
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
If reflux was necessary then I would have specified reflux. If you should need to add any makeup water, or to dislodge any crusting, just use
distillled water from a wash bottle as needed.
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
Yeah, so it looks like I was probably wrong about my picric acid from ASA being the cause of my problems. I tried another clathrate synthesis with
some very nice looking TNP from phenol, and guess what, I got basically the same result as the first time. Well I suppose I should be happy, because
if it was the cause, it would have just indicated another/different problem (one with the TNP).
Here is a picture of the ~8g of picric acid from phenol that is left after a couple of experiments. I put no effort or time into crystallization, as
the TNP was to be used just as a reagent.

I next moved on to titrating a solution of NaOH I made up. I measured out 8g of my NaOH and dissolved it in distilled water. Once dissolved the volume
was brought up so as to make (what should be) a solution of 4% NaOH by weight. An analyte was prepared from 96% sulfuric acid of trusted
concentration. One mL of the acid was measured using a graduated pipette, and dissolved in 50mL of distilled water. A drop or two of phenolphthalein
indicator solution was added to the analyte. The earlier prepared sodium hydroxide solution was added to the acid solution from a burette until a
persistent color change occurred. It was found that my 4% NaOH solution was actually closer to a 3.75% NaOH solution.
I decided to focus on Basic Lead Picrate until I get it figured out. If I can' t make the picrate, I can' t make the clathrate.
I used my titrated NaOH solution with known concentration, and tried a small Basic Lead Picrate synthesis. I did get a darker orange solution this
time. The picture shows a 50mL beaker with the orange lead picrate suspended in the reaction medium.

Here is a picture of what should have been basic lead picrate.

While better, I still don' t think it is dark enough. This leaves me with the lead nitrate to examine. Is it, or what it is possibly contaminated
with, adding more than normal acidity to the reaction and throwing off the pH?
I think you are right about the pH issue now. I now have a NaOH solution of known concentration, and I am fairly confident I have good TNP. How likely
is it that there is something wrong with my lead nitrate? I used lead chimney flashing to make it, if that matters.
Here is the basic lead picrate synthesis I used from the first page of patent 1,478,429. The whole patent was posted by Rosco in the thread "Picric
Acid Different Instruction".

[Edited on 16-8-2011 by Hennig Brand]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
That isn't nearly dark enough to be basic lead picrate. You need a tall form beaker with a deep swirling vortex sweeping the bottom and churning
everything in suspension. I don't think your reaction conditions are right.
Too small a scale is likely a big problem because you can't develop the vortex and get the suspension effect needed in that small of a beaker .....a
500ml or larger berzelius is needed and really a 1000ml might be even better. You basically want a tornado in a sleeve, and nothing duning on the
bottom. Let me check my notes for basic lead picrate
and see if I can tweak Friedrich's numbers for you.
The literature is correct and has been exhaustively verified. Basic lead picrate is orange brown like a dark rust color not quite as dark as lead
styphnate. You are correct to get the basic lead picrate right as a first step. Sheet lead is generally very pure lead, and it has to be pure so it
will be soft and easily formed and so that it will have good corrosion resistance. You could have residual acidity particularly if the nitrate salt
is not well dried. The order of addition must be correct and there must be sufficient time allowed for the reaction to continue to run with stirring
and heating being continued past the point of development of the precipitate, let it keep running twenty or thirty minutes at near boiling with
vigorous stirring after the end of addition. You may just be not letting the reaction run long enough. I'm sort of baffled at this because I've run
these reactions several times without any issue. I don't do anything extraordinary, just use pure ingredients as described and everything goes fine.
You could try this
Basic Lead Picrate
Experimental:
4.65 grams of picric acid is added to 100ml distilled H2O.
1.6 grams of NaOH is added to 35ml distilled H2O and upon dissolution is added to the previously prepared picric acid in water. When all is in
solution this alkaline sodium picrate solution is placed in an addition funnel.
7.3 grams Pb(NO3)2 is added to 100ml distilled H2O in a 500ml tall form beaker on a 7 X 7 Cimarec stirrer hotplate.
A polygon profile 1 X 3/8" stirbar is a minimum requirement.
The solution is heated with stirring to just below boiling point. Try to maintain a range of 95-98C and begin dropwise addition of the alkaline
sodium picrate at a rate of 1 drop every 2-3 seconds with stirring and heating continued for
at least ten minutes after the end of the addition. Yield is 9 grams basic lead picrate 98% of theory.
A note in the margin for a slightly different but similar experiment was made that most of the color deepening from bright yellow to much darker
bronze color occurs during the continuation of stirring and heating after the end of the addition, the final color development is a color described as
a "brick red coloration" reddish orange brown
very fine microcrystalline precipitate which is dense and settles instantly when stirring is stopped.
Different experiments used greater dilution for the lead nitrate solution up to 300ml and varied the amount of lead nitrate up to 8 grams, but yields
were lower at 8.5 grams
for a similar product.
So here's a song which seems appropo
http://www.youtube.com/watch?v=ccenFp_3kq8 I Can't Go For That (No Can Do) official video
http://www.youtube.com/watch?v=cGZwPGsfcwM audio only
[Edited on 16-8-2011 by Rosco Bodine]
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
Thanks for the analysis. I will try and focus on the procedure this time around, and do a full scale reaction. In the picture above I was only using
0.5g of picric acid in a 50mL beaker, so as not to waste chemicals, as you suggest this may have caused a big problem. By trying to save chemicals I
probably shot myself in the foot.
I will try a full scale basic lead picrate synthesis and use due diligence when following the procedure, using extra care when following temperatures,
rates of addition, etc.
[Edited on 16-8-2011 by Hennig Brand]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Scale matters for sure if it adversely affects the efficiency of suspension of the particles, as a circulation loop is essential for these reactions
to proceed, a high speed stirrer is needed here to effect what is something like a hotplate blender.
Usually if you set the beaker somewhat off center it will destabilize the vortex from being concentric axially oriented in a beaker, and if you are
lucky the vortex will do an obital sweep at a slow rate to cyclically churn up any duned particles. It is absolutely essential to maintain the
stirred suspension and that is difficult because the particles are dense, their tendency is to stay on the bottom against anything but a very strong
current of liquid which keeps them hydraulically mined from their little temporary hillsides being continually swept away. A centrifugal impeller
having a coaxial intake tube near the bottom to vacuum up the sediment and discharge it radially above would probably be the ideal setup.
http://www.youtube.com/watch?v=tkdqXfasg80 One On One
http://www.youtube.com/watch?v=D0LPNJUGnT8 Say It Isn't So
For the trained observer, What do you see in the final frame ?
[Edited on 16-8-2011 by Rosco Bodine]
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
Okay, I did a full scale basic lead picrate synthesis. I kept the temperature between 90 and 95C through the whole addition of the basic sodium
picrate solution. The addition was done painfully slowly taking about 2 hours to complete. The stirring was extremely vigorous using magnetic
stirring.
After the end of addition the stirring was continued for an additional 40 minutes with the temperature kept around 90-95C. After the 40 minutes the
heat was turned off, but stirring was continued as the beaker slowly cooled.
Here is a picture of some of my yield on a white piece of paper to the left of a small sample of the stuff made before in the small beaker. As you can
see it is darker but not that much. It is however much more crystalline and free flowing.

The only thing left that could be at fault is my lead nitrate(I think), but I don' t see any reason why it should be.
Could it be that some very subtle difference, possibly a minor contaminant could cause a drastic difference in color? Could my sample still be mostly
chemically pure? According to several internet sources lead styphnate for instance can be any color between yellow and brown.
At this point I am hoping I am going to be able to read something that will break the mystery.
BTW, my samples from the attempted azo-clathrate syntheses are very powerful primary explosives, even though not the right color.
Edit:
I have some reagent grade lead carbonate, I suppose I could use that and see if it made a difference. What do you think?
[Edited on 17-8-2011 by Hennig Brand]
|
|
|
quicksilver
International Hazard
Posts: 1820
Registered: 7-9-2005
Location: Inches from the keyboard....
Member Is Offline
Mood: ~-=SWINGS=-~
|
|
I had a lot of fun with this synthesis & had limited difficulty. However my basic lead picrate was a known good sample. I'm thinking about
Roscoe's comment re: scale because superficially I don't see any problem but the "vortex" is tough to maintain with any but a larger-level stir plate
and bar about 4". I was lucky as I have a very powerful plate and one bar that will get a damn fast swirl.
A long time back someone made a comment re: coloration and hydrogen. I tried to look for that as I thought it might be related (but no luck).
note: coloration (my sample) was a "sunshine yellow" to almost white-yellow, a bright crunchy crystalline of broken irregular shape w/ consistent
hardness.
[Edited on 17-8-2011 by quicksilver]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Actually the 7 X 7 Cimarec has a 2 pole motor that runs twice as fast as the 4 pole motor in the 12 X 12, and with a small stir bar running high speed
it will begin
an orbiting precession sort of effect that does a good bottom sweeping any dunes of solids off the bottom. I have a range of stirbars from microfleas
up to the humungus so I know the drill. It takes a bit of trial and error to find what combination is right for a particular situation.
What is to account for the light coloration I have no idea. That doesn't track with
what I have gotten which is a distinctly more bronze color. This isn't just my observation over the years, but pictures from a couple of other people
besides quicksilver have showed the bronze coloration also. I would say try pushing the temperature right up to the b.p. and backoff just slightly.
The reason I have run this reaction hotter is because I observed it worked better at the more elevated temperature. It still worked at a lower
temperature but went a lot slower.
The slowness of the addition is not so critical on the basic lead picrate stage, the run in of the addition over a half hour is adequately slow....at
least for the mid range dilution, because I have a note to the effect a half hour for the addition on one of the experiments I did. The really more
tediously slow addition is more important for the azide
when the basic lead picrate substrate is being further reacted.
What is the weight of the yield you get of the light colored product?
[Edited on 17-8-2011 by Rosco Bodine]
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
The yield for my last attempt, (from 2-3 days ago), at the azo-clathrate was 92% of your stated yield. I only started with 1g of picric acid however
instead of 4.6g. Here is a picture of the yield (4.8g), from one gram of picric acid. It is a free-flowing pile of tiny crystals. I may have used 1 or
2% for testing, and I may have lost 1% or so in the filter paper. So a 94-95% yield is probably a pretty good estimate actually. I should have weighed
it right away, instead of now when asked about it.

I got some things figured out today in regard to lead picrate as well. The color is all about the pH. The method controls the color a bit in the sense
that denser crystals of the same material will make the color more concentrated and appear darker. Here is a picture of the yields from the last few
attempts. I put much less care into the sythesis this time, I just added more NaOH. Which little pile do you think belongs to the last synthesis were
I raised the pH?

I increased the amount of NaOH by 40%, but kept everything else equal. I was still using the basic lead picrate method from that patent you posted in
the picric acid thread.
Just to be sure I titrated my NaOH solution again, but this time against hardware store HCl. I got a value that was different from the first time,
(against H2SO4), by only 0.4%. A difference which could easily be explained by the accuracy of my titration, I think. The hardware store acid is
probably pretty darn close to what it says it is.
[Edited on 18-8-2011 by Hennig Brand]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Now you are getting somewhere And yes it is a pH driven transition type
reaction....you gotta wait for it....and wait for it....but the color transition does and should happen.
What's that French word .....finesse 
As a trained observer, I would say the sample on the bottom right is
the genuine article ....
accept no substitutes.
http://www.youtube.com/watch?v=svAs-6MiqxE Aint Nothin' Like The Real Thing
http://www.youtube.com/watch?v=TOrBgJUC6gw Aint Nothin' Like The Real Thing
A factor involved which may cause some slight variance on the alkalinity is the progressive hydrolysis of the Pb(NO3)2
to the basic salt which does occur in hot water and the further that goes, the lower would be the requirement of NaOH, because there is some loss of
HNO3 which is volatile.
[Edited on 18-8-2011 by Rosco Bodine]
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
Holy crap, I just lit a little of my new basic lead picrate. Now it still isn' t as good as lead styphnate, but it is one hell of a lot closer to it
than any other lead picrate I have made thus far. Very neat.
BTW, if people are wondering why we are talking about lead picrate it is because it is a fundamental part of the clathrate synthesis. Divide and
conquer.
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Yeah the basic lead picrate is very close but not quite as energetic as lead styphnate, and has about the same nitrogen content. It really seems odd
how normal lead picrate having twice the nitrogen as either basic lead picrate or lead styphnate would not be as powerful, but even the anhydrous
normal lead picrate is a lesser beast than the other two ......go figure. However this observation is precisely what has made me wonder about the
possibility of a double salt of normal lead picrate and normal lead styphnate, wondering if that may result in yet another anomaly. Predicting what
may be the resulting performance is really hopeful guessing about these sorts of compositions.
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
Maybe its like the old expression ,"it's not what you've got, its what you can do with it". I am really impressed with the performance difference
between the two different forms of lead picrate. Now it is time to try the clathrate again, soon. I am just about out of lead nitrate though, except
for 3 or 4 grams. Maybe not tomorrow, but I see a clathrate experiment in my near future.
I suppose this double salt of normal lead picrate and normal lead styphnate isn' t something which shows up in the literature. It would be interesting
if we could get it to form, and do some testing on it.
A little off topic but, what is the performance difference between the two different lead styphnates, the normal and the basic?
[Edited on 18-8-2011 by Hennig Brand]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
I couldn't tell you about the difference between the normal and basic lead styphnate because I never made the basic lead styphnate. The idea I had
for the double salt of the normal lead picrate and the normal lead styphnate was that it could be a way of obtaining the anhydrous salt of each in
mixture as compared with the monohydrates which is the usual form. Anhydrous versions tend to be more powerful. You could try making the non-azide
clathrate from the basic lead picrate substrate. To do this leave the basic lead picrate stirring after it forms. Refill the addition funnel using a
second additional amount of lead nitrate solution a bit more than what was used first 8.5 grams Pb(NO3)2 , mixed with a slight excess molar amount of
potassium chlorate 2.8 grams KClO3 in about 80 - 100 ml distilled H2O and resume dropwise addition with continued heating and stirring as before.
Yield should be 14.6 grams 90.7% of theory of basic lead picrate - lead nitrate - lead chlorate triple salt / clathrate, mustard yellow prismatic
crystals having a strangely sparkling blue-purple silver glitter effect in sunlight. The crystals are parallelogram solids with all axes shifted,
like a cube twice shifted out of plumb both laterally and front to back. A perchlorate variant should be interesting also but has not been tried.
This triple salt is a particularly good absorber of lead azide and appears may possibly be able to contain up to 16 moles of inclusion absorbed /
caged lead azide. This triple clathrate is strictly "off label" and experimental and has not been made repeatedly or studied extensively for good
confirmation. Uncertainty and not having done analysis for confirmation causes me some reservation about identifying it as a 4/16 compound. It appears
to have a bit more sass than the 4/12 nitrate variant. The perchlorate variant is still one of those things on my "to do" list if / when time ever
allows.
[Edited on 18-8-2011 by Rosco Bodine]
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
I would like to try your non-azide triple salt/clathrate at some point for sure, but first I need to make more lead nitrate and figure out a few more
things. I tried another small batch of the azide clathrate by Mr. Anonymous using the last of my lead nitrate (5.5g), and once again to get the nice
dark orange color before the sodium azide addition I had to add a large excess of NaOH. After subtracting the amount of NaOH needed to neutralize the
picric acid I found the amount of excess NaOH needed was approximately proportional to the amount of lead nitrate used in the last two syntheses(basic
lead picrate and clathrate syntheses). My lead nitrate looks dry but it still must have a lot of excess acidity.
Well I think I have got most of my problems figured out. Sure as anything that was the main reason I was having all that trouble a year ago as well
when trying to make basic lead picrate. I assume you make your own lead nitrate. I am about to make another bunch. Any words of wisdom? I guess I
could do what I have been doing and then recrystallize it, or maybe just let it dry for an extended period of time.
Whatever I do, I don' t want the lead nitrate to get hydrolyzed very much.
[Edited on 18-8-2011 by Hennig Brand]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Keep in mind it is possible to form a dibasic or a tribasic lead compound in the same reaction mixture, so you must be precise with your base
referenced to the picric acid.
The easiest and most direct way of making pure lead nitrate is using HNO3 somewhat diluted maybe half strength dilution, as it attacks lead more
readily than does the concentrated acid if I recall correctly. Lead nitrate will precipitate from the cooling solution and is simply filtered out and
dried with gentle heating. Toxic Oxides of Nitrogen are produced during the process of lead dissolving in the nitric acid, so this is not a reaction
to be considered benign or done absent isolation and ventilation. Silver and mercury may similarly be dissolved by nitric acid. The lead salts
preparation thread should have several alternative methods.
You will see a visual indication of hydrolysis of lead nitrate in a near saturated solution because the basic salt is less soluble and will cloud the
solution or precipitate. A trace of cloudiness is then a good indicator of a lead nitrate solution that is nearly pure and neutral if it is strong
solution.
[Edited on 18-8-2011 by Rosco Bodine]
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
Thanks for the help. Just drying some fresh lead nitrate. I used 15% HNO3 because it was available as pH down. With the lead cut up fine and a little
heat, the process was actually quite quick (dissolved lead in ~15 minutes). I used gentle heating to get rid of ~3/4 of the water. I think I mentioned
that I had a bit of PbCO3, well I sprinkled enough in to neutralize most of the remaining HNO3. I am now drying the yield on a very expensive and
highly scientific drying apparatus (big piece of broken mirror).
I am going to try another small batch of basic lead picrate, and see if I have sorted out my lead nitrate problems. Then we tackle the clathrates.
Edit:
BTW, you mentioned something earlier that has kept me sort of interested. You mentioned that the basic lead picrate has half the nitrogen as the
normal lead picrate, but still is more powerful. I have noticed that explosive salts with heavy metal cations tend to be much more powerful than
explosive salts with lighter cations, but I don' t really know why. A sample of basic lead picrate has more of the heavy metal (lead) bonds, so I
guess that could be the reason for the greater power (not sure exactly why that is though).
Here are a couple pictures I found on the net. If they are wrong let me know. The first one should be normal lead picrate, and the second basic lead
picrate.
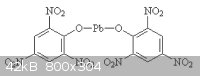 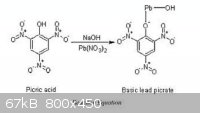
[Edited on 20-8-2011 by Hennig Brand]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Those structures appear to be correct. There would appear to be a relationship involving kinetic energy imparted to the "spectator ion" by the
explososphere components that is analogous to the specific impulse parameter associated with rocket propellants. Evidently there is a kind of mass
relationship matchup between the explososphere component and the spectator ion which is propelled by it and a good matchup results in an efficient
coupling for transfer of energy and resultant velocity being imparted to the spectator ion so that it behaves as a "bullet" being imparted a higher
velocity and kinetic energy. Some matchups between the explosophere and the spectator ion "bullet" are more efficient couples for the transfer of
energy than are other mass related or structure related matchups. And the arrangement in that regard is evidently more important with regards to the
net explosive effect than is a more general parameter such as "nitrogen content". Billiard balls scattering from impact
would seem to be a good analogy, where certain groupings arranged at specific angles transfer energy very efficiently but other groupings may
dissipate the energy. Calculating and predicting all the parameters involved there is above my pay grade. Nine ball in the corner pocket
|
|
|
quicksilver
International Hazard
Posts: 1820
Registered: 7-9-2005
Location: Inches from the keyboard....
Member Is Offline
Mood: ~-=SWINGS=-~
|
|
I have a poor memory for this but (if I AM correct) one of the contributors to clathrate research was an ordained Nun who had a remarkable background
in Chemistry. I MAY have a PDF somewhere with some of her work on lattice materials. It was one of the few bits and pieces that went beyond the
lead-oriented designs in the original patent. If I find it, I will post it.
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
| Quote: Originally posted by Hennig Brand | Yeah, so it looks like I was probably wrong about my picric acid from ASA being the cause of my problems. I tried another clathrate synthesis with
some very nice looking TNP from phenol, and guess what, I got basically the same result as the first time. Well I suppose I should be happy, because
if it was the cause, it would have just indicated another/different problem (one with the TNP).
Here is a picture of the ~8g of picric acid from phenol that is left after a couple of experiments.
I put no effort or time into crystallization, as the TNP was to be used just as a reagent.
http://www.sciencemadness.org/talk/files.php?pid=219088&...
I think you are right about the pH issue now. I now have a NaOH solution of known concentration, and I am fairly confident I have good
TNP. |
Hmmm....I have an idea which may be the mystery solver here about the pH issue. Did you recrystallize the picric acid from boiling water, or is that
8 gram sample on the filter what was gotten from dilution of the nitration mixture?
I know picric acid gotten from dilution of the nitration mixture usually has occluded acid from the nitration mixture and it doesn't take much of the
lower molecular weight acid as an impurity to substantially throw off the titration for what would be the usually required amount of base for
neutralization of *pure* (recrystallized) picric acid. You were needing to add more base so obviously you have acidity that is somewhere as a hidden
impurity, and I'm guessing it is in the picric acid if it wasn't recrystallized.
[Edited on 25-8-2011 by Rosco Bodine]
|
|
|
Hennig Brand
International Hazard
Posts: 1284
Registered: 7-6-2009
Member Is Offline
Mood: No Mood
|
|
I appreciate you doing an analysis of my problem. That wording doesn' t actually reflect what I actually did, and that is my fault. I was a little
unsure of how to say what had to be said right there. What I probably should have said was that no effort was made to form large, dense, gritty
crystals from the water of recrystallization because the TNP was to be used as a reagent and not an explosive.
I actually did recrystallize once from boiling water, and prior to that the crystals taken directly from the diluted nitration mixture were rinsed
well with small quantities of ice cold water. My recrystallized yield was close to 10g from 5g of phenol, which indicates a fairly strong/complete
nitration, I think.
I think my TNP from ASA was recrystallized twice, back when I originally made it.
The wording on my part was either not right, or open to interpretation.
I still think it is my lead nitrate that may have occluded acidity, if that is possible. Lots of people on this forum make lead nitrate, so if this
was the case one would think others must have had a similar problem at some point. I did notice the last time when making basic lead picrate, with the
lead nitrate I recently made, that I needed a much smaller excess of hydroxide. I still needed an excess, but it was much less.
[Edited on 1-9-2011 by Hennig Brand]
|
|
|
| Pages:
1
2
3
4 |








