Boffis
International Hazard

Posts: 1927
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Benzoin and related Acyloin via thiamine
Benzoin and related acyloin via thiamine catalysed dimerisation of aldehydes.
Benzoin and its related acyloins are important starting materials for the preparation of vicinal diketone (eg Benzil) and numerous organic ligands and
organic reagents used for the detection or analysis of metals, for example Benzoin-oxime and furil dioxime.
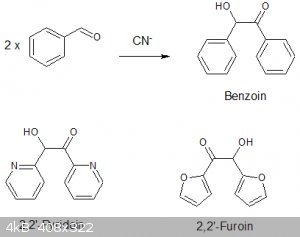
The preparation of benzoin from benzaldehyde via the alkali cyanide catalysed route is well know and has been investigated many times since its
discovery in the late 1830's. The problem for the amateur chemist is in part the availability of sodium and potassium cyanides but also the strongly
alkaline condition generated by the cyanide salts tends to promote side reaction such as Cannizzaro's reaction or tar formation with the more reactive
aldehydes.
Having attempted Vogel’s preparation of furoin (1) using sodium cyanide and finding the resulting material very difficult to purify and the yield
rather poor (about 23% in my hands) I decided to try the effect of thiamine on furfuraldehyde. Before trying this procedure on my precious
furfuraldehyde I decided to try it on the more readily available benzaldehyde. Most of the procedures I have found are for micro or semi-micro scale
preparations so I scaled up one of these procedures, an anonymous college procedure, that I found on the internet.
Benzoin from benzaldehyde via Thiamine nitrate:
To a 500ml round-bottom flask containing a 35mm stir-bar were added 7.034g of thiamine mono-nitrate and 20ml of water, the thiamine did not dissolve
completely. To the milky suspension were added 4ml of 40% sodium hydroxide solution and 75ml of rectified spirit (95% ethanol). The stirrer was turned
on and when the initial bright yellow colour had faded to a clear solution 39ml of benzaldehyde was added all at once. The flask was placed in a water
bath at 50°C and the temperature slowly raised to 60°C over about 15-20 minutes.
 Flask and stir bar Flask and stir bar
 Thiamine nitrate slurry Thiamine nitrate slurry
 Thiamine slurry turns yellow on NaOH addition Thiamine slurry turns yellow on NaOH addition
 Yellow colour faded to a clear solution Yellow colour faded to a clear solution
The flask was then maintained at 60°C for a period of 2 hours and allowed to cool a little before being poured into a 250ml beaker. Crystals formed
rapidly and when the mixture had cooled to room temperature it was chilled in the fridge to 5°C (for about 2 hours). The crystals were filtered at
the pump and washed with 50ml of 1:1 rectified spirit/water mixture, this removed most of the yellow colour from the cake. The filter cake was then
slurried into a further 50ml of the alcohol water mixture and again filtered at the pump, sucked as dry as possible and then turned out onto a watch
glass and dried. The cake was dried at 35°C for 24 hours and weighed 16.87g representing a yield of 41.6% of creamy white Benzoin. The Mp was
133-134.5°C a little below the published figure of 135-137°C. The melting point may be raised to close to the published value by recrystallization
from methanol, 10.5ml of pure methanol being required per gram of crude benzoin. This is not necessary if the benzoin is to be used for the
preparation of benzil.
 The cooling reaction mixture The cooling reaction mixture
 Final washed and dried benzoin product. Final washed and dried benzoin product.
Furoin from furfural via Thiamine nitrate:
This is basically the same as the previous procedure but with furfural in place of benzaldehyde. To a 500ml RB flask containing a 35mm stir-bar were
added 7.065g of thiamine mono-nitrate and 20ml of water. The stirrer was started and 75ml of rectified spirit and 4ml of 40% sodium hydroxide solution
were added. Once the transient yellow colour had faded 40ml (46.4g) of freshly prepared furfuraldehyde (Note 1) were added all at once, the reaction
mixture became dark brown. The flask was placed in a water bath at 50°C and maintained at this temperature for 1.5 hours (Note 2). After about 1 hour
a pale crystalline precipitate began to form and the reaction mixture became paler, eventually an amber colour. When the heat was turned off the
mixture was stirred for a further 10-15 minutes and then poured into a 250ml beaker and allowed to cool to room temperature, the flask was rinsed with
a little (perhaps 5-8ml) of rectified spirit. The mixture was finally chilled in the fridge overnight at 5°C. The mixture solidified but when stirred
liquefied, the slurry was filtered at the pump and the yellow cake washed with 1:1 rectified spirit and water until the runnings were colourless,
about 70ml were required (Note 3).
The filter cake was dried at about 35°C on a large watch glass for 24 hours. The crude almost pure white crystalline product weighed 33.57g (72.3% of
theory) and melted at 135-136.5°C which is close to the ideal melting point of 135-137°C. If it is felt necessary to recrystallize the furoin then
7ml of pure methanol per gram are used and recovery is usually about 89% without work-up of the filtrate.
 Filtering the furoin reaction mixture note the colour of the filtrate compared to the washed cake Filtering the furoin reaction mixture note the colour of the filtrate compared to the washed cake
 The furion filter cake being returned to the beaker for further washing The furion filter cake being returned to the beaker for further washing
 Crude furoin drying note creamy colour Crude furoin drying note creamy colour
 Recrystallised furoin Recrystallised furoin
Note 1 The furfuraldehyde was freshly steam distilled and extracted with ether, dried with a little magnesium sulphate and the ether evaporated. The
resulting furfuraldehyde was used as such without further distillation.
Note 2 Furfuraldehyde is more succeptable to the effects of alkalis than benzaldehyde and so a lower temperature was use in this initial attempt. The
reaction time was shortened slightly because after 1.5 hours the reaction mixture was already the thick slurry of crystals.
Note 3 In subsequent runs it was found better to disperse the furoin filter cake into about 100ml of 1:1 rectified spirit/water mixture, filter at the
pump and wash with 30-40ml of the same aqueous rectified spirit. This gave a pure white crystalline product that required no further purification for
most purposes.
Pyridoin
Pyridoin was prepared by essentially the same reaction but using pyridine-2-carboxaldehyde in place of furfuraldehyde.
Into a 250ml RB flask equipped with a magnetic stirrer bar were placed 7.01g of thiamine mono-nitrate, 20ml of water, 75ml of rectified ethanol and
4ml of 40% sodium hydroxide solution. The bright yellow suspension was stirred until the colour had almost faded and a clear solution obtained and
then 35ml (39.4g) of pyridine-2-carboxaldehyde added. The reaction mixture was heated to 60°C and maintained at that temperature for 1 hour in an oil
bath. Orange crystals began to separate after only 10 minutes or so and after an hour the slurry of crystals was so thick it was hard to stir.
The contents of the flask were poured into a 250ml beaker and the flask rinsed out with a little 50:50 rectified spirit and water. The orange slurry
was cooled to room temperature and then chilled overnight in the fridge. The slurry was filter at the pump (7 or 9cm Buchner) and washed with about
100ml of 50:50 rectified spirit and water, sucked as dry as possible and then dried on a large watch glass. The yield of crude pyridoin was 34.13g of
bright orange crystals or about 86%.
 Rapidly precipitating pyridoin after just 30 mins Rapidly precipitating pyridoin after just 30 mins
 Crude pyridoin filter cake Crude pyridoin filter cake
Recrystallizing the pyridoin proved problematic, 1gm of crude pyridoin requires almost 50ml of boiling methanol and only 0.48g were recovered without
work-up of the filtrate. In the end it was found more practical, though giving a less pure product, to stir the pyridoin into 150 to 200ml of
methanol, bring to the boil, cool and filter. The orange crystalline product is pure enough for most purposes such as the preparation of pyridil and
pyridoin derivatives.
 Recrystallised pyridoin Recrystallised pyridoin
Observation
The thiamine catalysed dimerisation of furfuraldehyde and pyridine-2-carboxaldehyde to the corresponding acyloin is a great improvement over the
cyanide method (and in the case of pyridoin the boron trifluoride route too), giving both better yields and much cleaner products.
Ethanol was used as the reaction medium as this was use in the original description but methanol and possibly isopropanol may also work equally well.
It was found to be very important to allow the thiamine salt to stand in contact with the sodium hydroxide until the bright yellow colour has almost
faded away before the addition of the aldehyde. Addition of the aldehyde too quickly greatly impairs the yield. The addition of more thiamine has only
a slight impact on the yield with the more reactive furfuraldehyde but doubling the amount of catalyst with benzaldehyde raised the yield to 56% with
the same reaction time. The same effect can probably be obtained by using longer reaction times but at the risk of greater side reactions.
Not all aromatic aldehydes will undergo a benzoin type condensation and I see no reason why the thiamine method should extend the applicability into
these aldehydes if the mechanism is basically similar.
Useful melting point data:
Benzoin 135-137°
Benzoin oxime several forms with different Mps
Benzoin thiosemicarbazone 179°
Furoin 137-139°
Furoin oxime 160-161°
Pyridoin 161°
References:
(1) A. I. Vogel Textbook of Practical Organic Chemistry 3rd ed 1956
The next part will be the oxidation of these acyloin to the corresponding "acylil".
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
Very nice writeup! Nice to see someone doing some research. I know this is a lot of work. 
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2894
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
Very nice illustrative pictures and beautiful clear report.
Why do you write that: "Not all aromatic aldehydes will undergo a benzoin type condensation "?
It would be nice if it worked with polynitrobenzaldehyde...
I dream of hexanitrobenzoin and hexanitrobenzil...then hexanitrobenzil dioxime (probably sensitive initiating salts) and furazan, or hydrazine
dérivatives (dihydrazones and diazines)   
I wonder if the same would work with thiophen or pyrole based aldehyds (thus furane with a S or NH instead of the O) ... and as an energetic material
lover, with tetrazole based aldehyd HN4C-CH=O... I can only dream of HN4C-CO-CO-CN4H (and dioximes, furazan and dihydrazone, diazines) and salts (Na,
K, Li, NH4, N2H5, HONH3, Ag, Pb, Hg, Cu, HNO3, HClO4, HC(NO2)3,...)
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
Fery
International Hazard
Posts: 1129
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
Into 250 ml flat bottom flask was put 14,4 g (42,7 mmol) thiamine chloride hydrochloride C₁₂H₁₇ClN₄OS * HCl, Molar Mass 337.27 g/mol.
20 ml H2O added, vit B1 dissolved completely into colorless solution.
Added cooled solution of 3,3 g NaOH dissolved in 8 ml H2O - dropwise in cold water bath (exothermic), color changed to yellow.
Added 120 ml 85,5 wt% isopropanol (ethanol usually used, but I have a lot of this 85% IPA so I tried to use it... it is not contamined with MEK +
denatonium unlike commercial denatured ethanols).
Stirbar added into the flask and the flask was put into aluminium pot with oil bath onto magnetic stirrer.
While stirring, 81,7 g benzaldehyde added (0,77 mol), Molar mass 106.12 g/mol. In my case the 2 liquids were not completely miscible so the stirring
was essential (I do not know how it is with ethanol, whether miscible or not).
It was stirred at highest possible RPM using magnetic stirrer in an oil bath, T inside flask kept at 60 C.
Cone adapter with screw thread with thermometer inserted to watch the T of the reaction, the thermometer hold in the adapter using rubber band (the
plastic cap removed) so the apparatus was not completely air tight but the amount of air entering was reduced to minimum (also only little of air in
the flask which was almost full).
Air oxygen causes oxidation of benzaldehyde to benzoic acid thus neutralizing NaOH / making salt of vit. B1 from the fee base. Also the benzaldehyde
used should be with as low of benzoic acid as possible (use pure benzaldehyde from yet unopened bottle or shake it with NaHCO3 solution / dry / vacuum
distill before use).
Back condenser was unnecessary as the reaction was below refluxing temperature. I used 85% IPA as I have few L of it and it could be used because
water content does not mind. Also the volume of solvent was slightly reduced in comparison with methods available over internet, this solvent
reduction allowed to react more benzaldehyde in the 250 ml FBF (using larger flask e.g. 500 ml with more of air oxygen would cause more benzaldehyde
oxidation to benzoic acid).
After 1,5 h the product started to suddenly massive crystallize out with evolution of heat and raising T to 70 C. No vapors generated, back condenser
not necessary.
After cooling in fridge to 4 C, the solid product sucked on sinter as dry as possible using water aspirator vacuum pump.
Vacuum interrupted and into the sintered funnel was added 4 C cold mixture of 50 ml water + 50 ml methanol and the cake was slurried, then again
sucked using water aspirator as dry as possible.
The wet product recrystallized from methanol, total volume of approx 900 ml (so somewhat little more than 800 ml of methanol used).
Cooled slowly to room temperature and then to 4 C in fridge overnight.
Crystals sucked on sinter, washed 2 x 50 ml of 4 C cold methanol on sinter and sucked as dry as possible.
Air dried for 1 day until the weight stable with no more decreasing.
m.p. 134-136 C
yield 48,2 g (0,45 mol = 58%) Molar mass 212.24 g/mol.
From mother liquor distilled out cca 750 ml of methanol, then remainder in distillation flask cooled down and 4,7 g second crop obtained (again sucked
out on sinter, washed 2 x 10 ml of 4 C cold methanol on the sinter). m.p. not determined, this second grade crop will be used in further syntheses
(benzoinoxime = Cupron indicator, oxidation to benzil and then diphenylglyxoime = Niclon 2 indicator and condensation of benzil with urea into
Phenytoin = antiepileptic drug syntheses)
total yield with second crop 52,9 g (0,50 mol = 65%)
Perhaps I got better yield thanks to low benzoic acid contamination and reducing air content in the reaction flask, perhaps only Vit B1 free base is
catalytic and its salts are less powerful?
thiamine chloride hydrochloride 14,4 g (catalyst)

vit B1 dissolved in 20 ml of H2O

after addition of cold solution of 3,3 g NaOH in 8 ml of H2O, dropwise, while stirring and cooling (exothermic) - free base of vit B1 is yellow

benzaldehyde 81,7 g for good yield it is necessary to be pure from benzoic acid and avoid air oxidation (benzoic acid would convert vit B1 free base
into salt with probably less catalytic activity)

it was necessary to stir the reaction as benzaldehyde did not completely mix with the reaction mixture (in my case with 120 ml of 85 wt% IPA, reduced
volume of solvent)

the reaction was kept stirring at 60 C in an oil bath on the magnetic stirrer (aluminium pot necessary or glass dish, any material which does not
shield magnetic field, or overhead stirrer could be used too)

after 1,5 h at 60 C and max RPM stirring the reaction suddenly started to crystallize out with slight T increasing from 60 to 70 C - I do not know
whether it was crystallization heat or reaction heat, refer to note 4 in this link, they observed the same: http://www.orgsyn.org/demo.aspx?prep=CV1P0094
 
sucked on sintered glass funnel, yellowish product
   
after washing with cold mixture of methanol with water 1:1 on the sinter, the product almost colorless, notice the dark mother liquor in the suction
flask

the crude wet product crystallized from methanol, the solvent was added in 100 ml increments, when total volume 800 ml the product did not yet
dissolve completely, dissolved completely when total volume 900 ml - as methanol is quite toxic, I suggest using RBF with back condenser, here I used
only a beaker with a RBF with cold water on top

crystals of the pure product

crystals filtered out on sinter, washed with cold methanol, later after drying 48,2 g of pure benzoin, m.p. 134-136 C

mother liquor, later I distilled out circa 750 ml of methanol from it and then the remainder in distillation flask was cooled, crystallized, second
grade product obtained (4,7 g) which m.p. was not determined, but which is still pure enough for further experiments (benzoin oxime = Cupron
indicator, oxidation to benzil, synthesis of benzil dioxime = diphenylglyoxime = Niclon 2 indicator, condensation of benzil with urea into
antiepileptic drug Phenytoin)

[Edited on 19-11-2021 by Fery]
|
|
|
|









