| Pages:
1
2
3
4 |
Nicodem
Super Moderator

Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
The reference about the influence of tertiary amines on the regioselectivity of the Reimer-Tiemann formylation of guiacol was already posted:
https://sciencemadness.org/talk/viewthread.php?tid=9596&...
The reference for the regioselectivity effect of cyclodextrin is also there (guiacol is one of the tested substrates).
Another reference says:
| Quote: | Abstract:
Refluxing 2-methoxyphenol with CHCl3 in EtOH in the presence of NaOH and Et3N gave 76.5% vanillin.
Huaxue Shiji, 15 (1993) 184, 138. |
|
|
|
dangator
Harmless
Posts: 8
Registered: 22-1-2008
Member Is Offline
Mood: No Mood
|
|
A nice trick for conjugate reduction of the enone formed after the aldol condensation/dehydration is CuI/LAH. See Tet Let No. 50 4453-4456 or I can
mail it to whomever needs it.
|
|
|
solo
International Hazard
Posts: 3967
Registered: 9-12-2002
Location: Estados Unidos de La Republica Mexicana
Member Is Offline
Mood: ....getting old and drowning in a sea of knowledge
|
|
The most recent issue of Tetrahedron Letters is Volume 49, Issue 7, Pages 1091-1282 (11 February 2008), so tell me how you propose to provide a
reference that doesn't exist.......also why didn't you just upload it, whatever you have as a reference surely isn't the reference
noted................solo
It's better to die on your feet, than live on your knees....Emiliano Zapata.
|
|
|
dangator
Harmless
Posts: 8
Registered: 22-1-2008
Member Is Offline
Mood: No Mood
|
|
Lol @ internet tough guy.
Attachment: lahcui.PDF (163kB)
This file has been downloaded 1281 times
|
|
|
not_important
International Hazard
Posts: 3873
Registered: 21-7-2006
Member Is Offline
Mood: No Mood
|
|
Volume 16, Issue 50, not Number 50. Volume 49, Issue 7 is the current one.
Also in JOC:
New and effective reagents for 1,4 reduction of .alpha.,.beta.-unsaturated ketones, LiAlH4-CuI and Its reactive species H2AlI
E. C. Ashby, J. J. Lin, and R. Kovar
Journal of Organic Chemistry
Vol. 41, No. 11: May 28, 1976
pp 1939 - 1943; DOI: 10.1021/jo00873a011
|
|
|
solo
International Hazard
Posts: 3967
Registered: 9-12-2002
Location: Estados Unidos de La Republica Mexicana
Member Is Offline
Mood: ....getting old and drowning in a sea of knowledge
|
|
Reference Information
New and effective reagents for 1,4 reduction of .alpha.,.beta.-unsaturated ketones, LiAlH4-CuI and Its reactive species H2AlI
E. C. Ashby, J. J. Lin, and R. Kovar
Journal of Organic Chemistry Vol. 41, No. 11: May 28, 1976, pp 1939 - 1943;
Attachment: New and effective reagents for 1,4 reduction of .alpha.,.beta.-unsaturated ketones, LiAlH4-CuI and Its reactive species (551kB)
This file has been downloaded 1911 times
It's better to die on your feet, than live on your knees....Emiliano Zapata.
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
LiALH4 is a bit of an overkill for such a reaction IMHO. If the reduction can be easily done with cheaper, safer reagents, such as NABH4 and
H2/Catalysts, using expensive LiALH4 is a bit of waste. But of course that simply depends on your financial aids/work environement  . .
I'm sure there's lots more one can do with LiAlH4 that can't be done by other means. Oh well, just another article in another journal I guess.
BTW, the two recrysatllized crops had a sharp 127°C melting point (lit: 127–128.5 °C ) , so I think I can safely say it quite pur. The crystals
have a lovely, light vanillin-like smell, a bit more fruity.
|
|
|
LSD25
Hazard to Others
Posts: 239
Registered: 29-11-2007
Member Is Offline
Mood: Psychotic (Who said that? I know you're there...)
|
|
Klute,
Here is a Pd-CTH enantioselective reduction of a-methylcinnamic acid to the corresponding (S)-2-methyl-3-phenylpropanoic acid...
It proceeds at fairly low temps, seems remarkably simple (in fact seems awfully like, well apart from the time, the one I posted earlier - albeit it
is CTH).
I'd dearly love to know how the l-tartaric acid promotes the claimed huge increase in ee's (98% ee or 99% of the (S))
PS Thanks Stoichemetric Steve for this site
Whhhoooppps, that sure didn't work
|
|
|
Filemon
Hazard to Others
Posts: 126
Registered: 26-4-2006
Member Is Offline
Mood: No Mood
|
|
You may make it condensation with p-hydroxibenzaldehyde and acetone => p-hydroxibenzalacetone
You reduce p-hydroxibenzalacetone with Zn + AcOH.
http://www.orgsyn.org/orgsyn/pdfs/CV1P0077.pdf
http://www.orgsyn.org/orgsyn/pdfs/CV9P0186.pdf
|
|
|
carbonic
Harmless
Posts: 17
Registered: 22-2-2008
Member Is Offline
Mood: No Mood
|
|
This is truly interesting and informative post! A nice change from all the posts on aminating questionable substances and so on...
I hear a lot about the thermogenic power of Raspberry ketone - just a little bit will make you warmer immediately, and it is very popular with the
weightlifting guys these days.
So rhodium is out of the question for me, anyone can suggest a simple and gentle hydrogenation that is a little more OTC? pd/c is too strong? And
nobody sells borohydride (even for fuel cell) anymore!
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Yes, apparently Pd/C has been often employed for such reduction, although the problem is a certain amount of the saturated alcohol is also formed, and
the ketone and alcohol are hard to effectively seperate. A carefull coluum chromatography could do the trick, or maybe a bisulfite if the adduct isn't
too soluble...
I have still not proceeded to any hydrogenations, although I now have sevral unsaturated ketone at hand: straight phenyl, 3-MeO-4-OH; 2-MeO;
3,4-MeO, etc. Most of the unsaturated ketones have a light, pleasant smell.
Raney nickel is said to work for such reduction too. I'm surprised you can't find any NaBH4, it is relatively availble.
|
|
|
LSD25
Hazard to Others
Posts: 239
Registered: 29-11-2007
Member Is Offline
Mood: Psychotic (Who said that? I know you're there...)
|
|
Is it possible to just use crushed-up automotive catalytic converters as is? They are supposed to contain 'some' rhodium, in addition to slightly
larger quantities of both platinum and palladium. As they are designed to be catalysts per se, would they not be able to be utilised? IIRC the
platinum group metals are supported on predominantly alumina, which shouldn't cause too much trouble... (although I am unsure if the cesium oxide
content would be problematic).
Whhhoooppps, that sure didn't work
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Honestly, I can't say. The quality of the catalyst, impurities, catalyst poisons, support, ect all play a very important role, for example home-made
Pd/C never has the same activity as commercial catalyst. It could be interesting to try, but I would use a "simplier" substarte, one where only 1
reduction product is possible, and easily seperated from the substrate for such experimentations. This reaction already seems delicate with commercial
catalyst, the result of using such material wouldn't bring any reliable information IMHO.
|
|
|
carbonic
Harmless
Posts: 17
Registered: 22-2-2008
Member Is Offline
Mood: No Mood
|
|
Used cat. converters are poisoned already by exhaust. So in addition to the catalyst being deactivated you are going to introduce all the heavy
metals from 10yrs of gasoline.
Now my warning is done, so I will show you someone else recycling cat. converters. But he is simply using it to degrade CO as it was intended.
However, it supports your recycling scheme.
http://mattson.creighton.edu/CO/index.html
|
|
|
not_important
International Hazard
Posts: 3873
Registered: 21-7-2006
Member Is Offline
Mood: No Mood
|
|
| Quote: | Originally posted by LSD25
Is it possible to just use crushed-up automotive catalytic converters as is? They are supposed to contain 'some' rhodium, in addition to slightly
larger quantities of both platinum and palladium. ... |
Some do have rhodium, some don't. But they are optimized for their job, high temperatures and IC engine exhaust. You'd likely have to strip the
metals off the substrate and work that solution up to make a catalyst.
You might look into pentacyanocobaltate catalysts, which can be used to reduce double bonds in conjugated unsaturated but do not catalyse reduction
of isolated C=C double bonds. While normally used on dienes and similar hydrocarbons, I dimly remember it being used with unsaturated ketones.
|
|
|
Fleaker
International Hazard
Posts: 1252
Registered: 19-6-2005
Member Is Offline
Mood: nucleophilic
|
|
Very nice post Klute.
Many catalytic converters contain rhodium along with other amounts. I doubt that they would be poisoned, but they will definitely not be as effective
as freshly prepared catalysts done conventionally with BH4- onto carbon or alumina.
LSD25, it's cerium oxide, not cesium mixed with the gamma alumina substrate.
Neither flask nor beaker.
"Kid, you don't even know just what you don't know. "
--The Dark Lord Sauron
|
|
|
LSD25
Hazard to Others
Posts: 239
Registered: 29-11-2007
Member Is Offline
Mood: Psychotic (Who said that? I know you're there...)
|
|
ooops, did I really write cesium? I did mean cerium - I have been playing with 'em for three weeks now, the trouble is that my only source of sodium
nitrite is out at present. This means I have to try and find a good basic resin, any ideas (I have been looking into the ones used for water
deionization not the ones for softening they are acidic)
PS Are the exchange resins used in aquaria the same as the ones we want here, the ones that remove chlorine from aquarium water?
Whhhoooppps, that sure didn't work
|
|
|
LSD25
Hazard to Others
Posts: 239
Registered: 29-11-2007
Member Is Offline
Mood: Psychotic (Who said that? I know you're there...)
|
|
Sorry, I posted something which Solo had already posted (thought that sucker looked familiar).
BTW I recently encountered a silver oxide mediated decarboxylation-deamination of phenylalanine which reportedly gave mainly benzaldehyde (thus, it
'MIGHT' be useable to form the p-OH-benzaldehyde from tyrosine).
[Edited on 5-6-2008 by LSD25]
Whhhoooppps, that sure didn't work
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
I've FINALLY decided on trying to reduce the double bond on the unsaturated zingerone ketone.
I thought it would be a good occasion to start working with atmospheric catalytic hydrogenations. I decided on starting with aregular hydrogenation
over recycled Pd/C, and see what gives, before trying out cheaper catalyst, so than I coul dcompare yields, hydrogen uptake rates, ratio
ketone:alcohol, etc
I tried a setup using a chromatography column as H2 reservoir, but apparently it didn't work well as the pressure must diminsih too much after a
little absorption to let the recation going.
I also did a stupid mistake, adding K2CO3 to a phenol w<hile I didn't want the phenoalte...
Synthesis of Zingerone 4-(4-hydroxy-3-methoxyphenyl)butan-2-one by atmospheric catalytic hydrogenation over Pd/C
Ref: Us patent N° 5,847,225
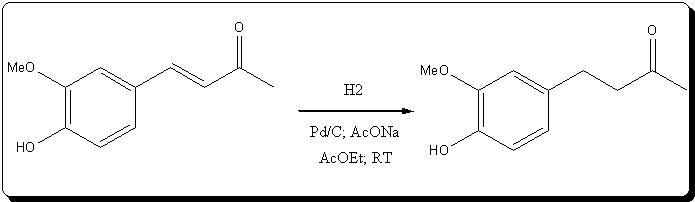
The following setup was assembled:
A 100mL schlenk was connected via a gas outlet adaptator and plastic tubing to a 350mL chromatography column mounted with a schlenk tap. The bottom
of the column was immersed in a beaker containing 500mL water, the column's tap fully opened.
A hydrogen generator was assembled: a 250mL 4-neck RBF with a stir bar was mounted with a 70mL addition funnel, itself mounted with a gas outlet
adaptator connected to some tubing.
The column:

The complete setup:

40mg of recycled Pd/C from a previous CTH reduction was charged into the schlenk, along with 10mL AcOEt.
The setup was then charged with hydrogen in the following way:
A very weak vacuum was connected to the schlenk's side arm. The schlenk tap was opened, aswell as the tap on top of the column. The water started
rising into the column. Once it was near the top, the tap on top of the column was closed, followed by the schlenk's side arm. The vacuum tubing was
disconnected, and the argon tubing tubing attached after purging the side arm a few times (attach the tubing, leave some pressure to build up, briefly
discconect the tubing to evacuate the argon/air pressure, etc etc to chase any air from the side arm), then the schlenk tap was opened, followed by
the column. The water level fell, and a flow of argon was left to bubble through the setup for a few minutes.
The addition funnel was charged with 20mL 37% H2SO4, and 0.5g of zinc dust was slurried into the RBF with a little dH2O.
The schenk and column were once again evacuated until water level was near the top of the column, acid dripped onto the zinc, and a flow of H2 was
left to purge the generator for a min. The closed schlenk side arm was then purged with H2 as previously, the tap opened, and the column opened. The
column was filled with H2, and a flow of H2 bubbled through the setup until evolution of H2 slowed down. The column was sealed, filled with H2, and
stirring continued for 10min [Note1].
Catalyst during saturation:

The H2 tubing was disconnected, the schlenk briefly evacuated (column still closed), and flushed with argon [Note2]. The schlenk's top was
disconnected, and under a flow of argon, 0.39g (2.82 mmol, 0.25eq) of K2CO3 were added [Note3] , followed by a solution of 2.15g (11.20mmol)
of 4-(4-hydroxy-3-methoxyphenyl)but-3-en-2-one in 20mL AcOEt [Note4]. The erlen containing the yellow solution was rinsed with 5mL AcOEt.
The unsaturated ketone:

The obtained reaction medium:

the schlenk was closed, evacuated, back filled with argon, evacuated, back filled with H2 (adding more zinc dust to the H2 setup) twice, then the
schlenk arm was closed, the column opened, and vigorous stirring started. H2 uptake was immediate, but slow. Slight warming was noticed.

After 30min, the reaction medium had turned to a suspension of guacamole-green solid along with the catalyst: the phenolate. Ihad completly forgotten
there was a phenol group in the molecule, and that K2CO3 would form the phenolate... Even if this wasn't obviously a problem for the reduction, the
solid could cover the catalyst enough to limit H2 uptake, considering the weak loading. So 0.3mL (~5.5mmol) GAA were added (opening the schlenk after
performing all the operations described previously), along with a spatula (~1g) of dry AcONa. The green solid dissolved immediatly, leaving a faintly
emeraude-green solution and a few orange carbonate particules.
The phenolate:

After adding the acid:


It was noticed that the water level never went above a certain point, even after recharging the column with H2, more absorption occured, but each time
the level stopped at the same level (pretty low) [Note5].
It was decided to change the setup (not more than 30mL H2 absorbed over 3h) to a conventional inverted cylinder.
The flask was evacuated, aswell as the column, and back filled with argon. The column was disconnected, schlenk closed, and under a flow of argon the
gas tap was attached on the top of the schlenk. A tubing was attached from the top of this tap, and placed in the cylinder, which was inverted under
the surface of the water in the beaker. The setup was evacuated to rise empty the column from it's air, just under the tubing, and back filled with
argon, doing this twice.
Setup with inverted cylinder:


More zinc was added to the H2 generator, and the setup purged with H2 before filling the column. The schlenk side arm was then closed, and vigorous
stirring started. H2 uptake started immediatly, monitering the rate: 1mL/min.
After 20min, it seemed the H2 uptake has ceased. a White inorganic solid had precipitated, and seemed to cover the catalyst....
The inorganic precipitate on the catatalyst (very quick decantation):

The setup was opened (following the directions given previously, and 20mL MeOH containing 1 mL dH2O added. This completely dissolved the solids, but
H2 uptake still didn't continue. A spatula tip of potassium formate was then added: there was a slight gas evolution (column closed, side arm opened)
for a few minutes, that died off quickly. The column was then opened again after flushing the schlenkwith H2, and H2 uptake started immediatly,
quickier than before (2mL/min), and calmed down to 1mL/min over 15min [Note6].
Inorganic dissolved:

The absorption rate then gradually diminished (1mL/2-3min), but didn't stop. The hydrogenation was stopped overnight (sealed column, stopped
stirring), and restarted in the morning. The green color had lightened up considerably, and TLC (Eluant: 10:1 DCM:AcOEt) indicated a good amount of
saturated ketone was formed, along with a little alcohol [Note7].
Reation medium in the morning:

125mL H2 had been absorbed since the cylinder setup was assembled. I considered that roughly 40mL had been absorbed with the column, so there isn't
much left before reaction is finished, but I prefer following the reaction with TLC.
H2 absorption:

Each sample is taken after stopping the stirring and leaving the catalyst enough time to settle, and 0.2mL of the green solution are taken, placed in
a vial, and acidified with a drop of GAA. The first sample taken (1H afetr beginning of hydrogenation with the column) stayed green afetr
acidification, and only the unsat ketone was detectable by TLC. The sample taken 1h after switching to the inverted cylinder setup cleared up^entirely
after acidification, and showed 3 spots (see Note 7). Further samples show that the unsat ketone spots is diminishing, the sat ketone getting larger,
and the alcohol staying roughly the same.
TLC samples after acidification:

After a total of 24H hydrogenation, TLC indicated all the unsaturated ketone was consumed, and only a minimal amount of alcohol was present.
After leaving the catalyst decant, it was obvious the solution was completly clear.

The solution was vacuum filtered over 2 filters, the caatlyst washed with 2x5mL AcOEt and 5mL MeOH, and the clear filtrate transfered to a seperating
funnel. 50mL of brine was added, and the funnel gently shaken. The very slightly yellow organic was seperated, and the aq. extarcted with 2x10mL
AcOEt.

The combined organics were washed with 50mL brine, and dried 20min over Na2SO4. The solevnt was then removed under avcuum at 40°C, to afford a
clear pale yellow oil with a very pleasant smell.

The oil was placed in the fridge, covered with a little pet ether, to try and induce cristn, without succes
The crude product will be purified by column chromatography, or by bisulfite adduct formation. Details to come when done.
Notes:
Note 1: The was done to fill the column with hydrogen (250mL required to complete reaction), and to pre-saturate the catalyst with H2.
Note 2: Each time the setup was opened, it was evacuated and flushed with argon to chase any H2, to avoid any fire hazard.
Note 3: the base was added to hinder reduction to the alcohol. Unfortunaly, an acetate should have been used to not form the phenolate.
Note 4: Slight warming was required to acheive complete dissolution.
Note 5: The pressure needs to diminish considerably for the water leevl to rise up more than this level I suppose. maybe at a certain pressure the
equilibrium constant diminishes enough to stop the reaction. Prhaps the water leevl rising was simply du to leakes, although the joints were greased
and double checked, I decided to follow conventional directions (inverted cylinder), as these were sure to work.
Note 6: apparently formate can reactivate Pd/C catalyst efficiently. this measn saturating Pd/C before a CTH is useless, but adding a little formate
before a catalytic hydrogenation could substitue to conventional H2 saturation (no need to open the setup with H2-saturated catalyst: less fire
hazard)
Note 7: a few eluants were tried and this variation afforded perfect seperation: from bottom to top: the alcohol as a thin rim, the unsaturated ketone
(compared to pur sample), and then the saturated ketone. Rfs to come shortly.
Comments
Well, the selectivity of Pd/C is very encouraging, contamination with the amcohol is only minor by TLC, but nevertheless enough to prevent cristn.
On the other hand, the recation is much too long. Is it the low loading? The fact that's it's a recycled catalyst? I haven't tried any used catalyst
for other recations up to now, maybe the regeneration method isn't that efficient... I will see with another reaction.
I still plan on trying out other catalysts. I'm particularily interested by the Cu/SiO2, as it can be easily and cheaply made at home. On the other
hand, the authors of the patent use fumed silica, which I don't have. I will surely try chromatography silica (60-200 mesh, acros) and home-made
silica (less porous, but of varying quality).
I still have 2.15g of dehydrozingerone to play with, and enough vanilline to make some more (considering how easy it is..), but I would like to
obtain some 4-hydroxybenzaldehyde. I definitively can't find some at a decent price, and am out of phenol. I'm afraid I will not be able to buy some
until a little time,a s I've been spending too much lately and have other
projects to attend to (TEMPO derivatives, etc).
If anyone find a relatively cheap source of 4-hydroxybenzaldheyde, please contact me! I would be delighted to obtain ~50gr at a reasonable price.. I'm sure we could work out a compassion
[Edited on 16-6-2008 by Klute]
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
Great contribution! 
(Though I have to admit I have not read it all since I don't have much time at the moment)
| Quote: | Originally posted by Klute
The crude product will be purified by column chromatography, or by bisulfite adduct formation. Details to come when done.
|
Just a quick advice before you waste time with a chromatographic column. 4-Arylbutan-2-ones do form bisulfite adducts, but in my experience they do so
terribly slow and quite inefficiently. Use a threefold excess of NaHSO3 (or more) in aq. ethanol and let it stir for several hours at 40-60°C.
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
LSD25
Hazard to Others
Posts: 239
Registered: 29-11-2007
Member Is Offline
Mood: Psychotic (Who said that? I know you're there...)
|
|
As to the p-hydroxyphenol - anise/fennel essential oil ain't expensive and here's the route:
http://wmw.justice.gov/dea/programs/forensicsci/microgram/jo... (the link don't work, good)
| Quote: | Anisaldehyde (4-Methoxybenzaldehyde [2])
A freshly prepared and stirred solution of 30 mL concentrated sulfuric acid in 150 mL water was allowed to cool down to 30° C, and anise oil (9.8 g)
was added. A total of 25 g sodium bichromate was then added, at such a rate that the reaction temperature remained between 35-40° C. The reaction
mixture was extracted four times with toluene (75 mL each), and the combined organic phases were washed twice with 5 % NaOH (100 mL each), and once
with water (100 mL). The organic phase was evaporated to about 20 mL, and anisaldehyde was then isolated as its bisulfite adduct. The yellow
precipitate was washed with an EtOH/ether (1:1) mixture until the precipitate's color turned white (that is, similar to the bisulfite adduct generated
from commercially available anisaldehyde). Setting the anisaldehyde free resulted in 4.9 g of a yellow oil with a pleasant odor. The mass spectrum was
in agreement with an authentic sample. Anisaldehyde was the main product (95 % by GC/MS), but several minor impurities (not further identified in this
report) were noted. |
Still needs demethylating, but comparatively quick and easy
Whhhoooppps, that sure didn't work
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Thank you for your comments, both of you.
Nicodem, thank dfor the advice regarding the bisulfite adduct, especially I would have though it woulo dhave reacted pretty well being a methyl ketone
but... I guess I'll just do a quick column: I am one the rare chemists at work that actually LIKES doing columns 
Alice, that synth looks pretty nice, but I don't have any dichromates and am not particularily found of using them. But the degradation of tyrosine
with TCCA (IIRC?) that you proposed looks very interesting. If I can find some tyrosine locally, I wil give it a try.
At one moment I will have to get some more phenol, as I'm out of salicylaldehyde (had problems forming the N-ethyldiethylenetriamine ligand), so I
will try the Riemmer-Tiemann with triethylamine..
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
LSD25
Hazard to Others
Posts: 239
Registered: 29-11-2007
Member Is Offline
Mood: Psychotic (Who said that? I know you're there...)
|
|
Klute, I am on fucking drugs I swear - I meant to write p-methoxybenzaldheyde (anisaldehyde) not p-methoxyphenol
I also meant to say fucking nice job
PS Wouldn't the permanganate oxidation of anethole give the same result?
http://www.erowid.org/archive/rhodium/pdf/isosafrole2piperon...
http://designer-drugs.com/pte/12.162.180.114/dcd/chemistry/b...
The tyrosine variant uses Silver oxide as the oxidant which is going to make it too expensive.
PPS I also added something which Solo only just got today, the demethylation of substituted porphyrins with anilinium chloride, used in preference to
the more usual pyridine.HCl, HBr, etc.
[Edited on 17-6-2008 by LSD25]
Attachment: Demethylation.AniliniumChloride.pdf (210kB)
This file has been downloaded 1367 times
Whhhoooppps, that sure didn't work
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
After purification on a short column, using 50:50 AcOEtet Ether, 1.61g (8.30
mmol) of practically colorless zingerone was obtained, giving a 74.11% yield from the unsaturated ketone, and
59.18% from vanillin.
The oil is practically pur by TLC, only a very faint spot of the alcohol from the last fraction, but nevertheless is very hard to cristallize; it is
very slowly starting to cristallize in the freezer, with 2 nucleation points, very slowly propagating (over several hours). They look like mold
growing in the oil
The compound has a very pleasant, slightly pungeant though faint smell of ginger. Hurray!
Hopefully Cu/SiO2 will be more selective than Pd/C, and there will be no need of seperating the ketone from the alcohol, and it will possibly
cristallize more readily.
EDIt: I forgot to add the Rf's:
Eluant: 10:1 DCM:AcOEt
Saturated ketone: 0.65
Unsaturated ketone: 0.56
Alcohol: 0.45
[Edited on 18-6-2008 by Klute]
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
An update:
After considering the Orgsyn prep Filemon indicated, and the notes in OrgSyn preparation of benzylacetophenone, I decided to try reducing the
unsaturated ketones with Zn dust: the procedure seemed ridiculousy simple, and sued very easily accesable reagents. This would really make the method
of choice for preparation of phenolic phenylbutanones...
I gathered a few old Ber. refs on the reduction of bezalacetone and benzalacetophenone to their saturated ketones, and with the unvaluable
help of Woelen at translating german, managed to figure out the sparse details furnished... basicly, the zinc dust was added to the unsaturated ketone
in GAA, the mixture heated to reflux for 3-4H, the solids filtered off, GAA diluted with water,a nd products extarcted with ether.
Now, the Zn reduction of a,b-unsaturated ketones is said to produce varying amounts of a dimer, and higher polymers, which are only slightly soluble
in ether. This is with the non-phenolic unsaturated ketones...
I performed a few reductions of 4-(4'-hydroxyphenyl)but-3-en-2-one, here are my notes and some pics:
Preparation of 4-(4'-hydroxyphenyl)but-3-en-2-one
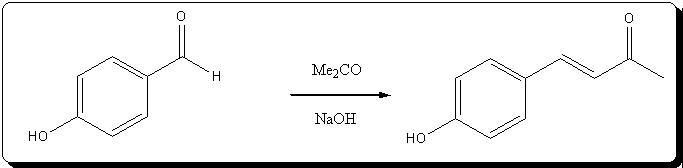
(no pictures, but basicly very similar to dehydrozingerone)
5,0 g (40.94 mmol) of p-hydroxybenzaldehyde are dissolved in 20mL (272.04 mmol) freshly distilled acetone inside a colorless 100mL
bottle, giving a dark orange solution, with a very small amount of some fine amber solids in suspension.
2,0g (50 mmol) of NaOH are dissolved in 20mL dH2O (10% w/w; 2.5N), and the cooled solution is slowly added to the stirred aldehyde solution.
At first, a beige voluminous solid is formed, but it quickly dissolves to give a very light yellow limpid solution, gradually darkening to a deep
orange.
The solution turns dark orange/brown over 1H, and is left to stand for 24H, with occasional stirring. A dark red solid appears after two hours, giving
a dense cristallin slurry.
The mixture was then added to 100mL of 10% HCl in a 250mL beaker, with good stirring. White fumes are evolved, and the dark orange/brown mixture warms
up. the beaker is cooled in a ice water bath with vigorous stirring, and the oily material crystallizes as a dark brown solid after a couple of
minutes. The suspension is stirred for another 10min at 5°C, then vacuum filtered over a glass frit. The brown mass is washed with a little amount of
ice-cold water, giving a dark brown filtrate and light green cristillin solid. The solid is dried by suction for 20min, and dried in a CaCl2
dessicator over night.
the solids are added to a 100mL erlenmeyer, and covered with 25mL toluene. The toluene is heated to ~80°C on a hotplate, and AcOEt added dropwise
until total dissolution of the solids (2-3mL required). An equal volume of toluene is added, and the dark green mixture is left to cool slowly. The
next morning, beautifull yellow prisms are obtained. The crystals are vacuum filtered, washed with cold toluene:AcOEt 10:1, and pet ether, and dried
by suction.
After 24h drying in a desicator, the crystals weighed 4.9g (30.21 mmol), 73.79%, and pur by TLC (single spot, eluant 3:1
AcOEtet ether).
By evaporating part of the mother liquors and adding pet ether, some more orange solids are obtained. They are recrysatllized by toluene/AcOEt to
offer 1.13g (6.97 mmol) of TLC pur yellow crystals, bringing the yield to 90.81%.

Notes:
-The first few times I left the reaction proceed for 48H, but it seems the yields are even better with 24h reaction time as they are less impurites.
-Dissolving the crude solid in a solvent, and washing with cold brine (the unsaturated ketone is sparingly soluble in H2O) could be a more efficient
approach.
-The recrystallization system is really adequate, I had trouble finding a good solvent system as the product kept on oiling out of solution with
EtOH/H2O, AcOEt/Pet ether, etc. Having the crude product as clean as possible gives better results.
-the product has a very slight, pleasant smell, much lighter than dehydrozingerone (which gives 80-90% yields by following this procedure using 5.0g
vanillin, all other reagents in same quantities)
Synthesis of 4-(4'-hydroxyphenyl)butan-2-one (Rheosmin)

1.62g (10.00 mmol) of the unsaturated ketone previously prepared were crushed to a fine powder and dissolved in 30mL GAA in a 100mL 3-neck
14/19 RBF, equipped with a CaCl2-guarded condenser, a thermometer and amgnetic stirring.


1.32g (20.00 mmol) of zinc dust were weighed out, and added in small portions to the stirred solution over 30min, at room temp. There was no
exothermic reaction of noticeable bubbling, and the yellow solution took a greenish tint before enough zinc was added to give a light grey suspension.


20min after addition:

The mixture was then heated to 50-60°C in an oil bath, gentle H2 evolution started, and the green/yellow color disappeared in 10min.
40min after addition (leaving the zinc decant before taking picture):

1H:

2H:

after 2h reaction time, the flask was cooled, and the unreacted zinc was filtered on a glass frit. the zinc was thoroughly washed with 2x10mL GAA.


The slightly yellow, cloudy filtrate was then added to 150mL ice-cold water, in a beaker immersed in a ice bath. The mixture immediatly turned pink,
and some ice was added.

Unfortunaly, a viscous semi-solid was obtained, but smelling very strongly of raspberries. Solid NaCl was added to precipitate even more of this
viscous paste.

The paste was dissolved in a small volume of MeOH, NaCl filtered off, and solvent evaporated. Pet ether boils on the obtained viscous liquid didn't
yeild any cristillin product. The slightly pink aqueous mixture was extarcted with AcOEt and the solvent evaporated afetr been washed with brine,
togive a viscous, raspberry-smelling paste. Attempts to cristallize this with various solvents and conditiosn were all fruitless.. A column
chroamtography could be done, but th eobjective is to find a easy reduction with acceable reagents giving a pur enough product..
TLC (3:1 AcOEtet ether) indicated saturated ketone (major stain), a little unsat
ketone, and two other spots (dimers).
Comments:
I performed a first reduction using 4x zinc, but directly extracting the diluted AcOH solution. After washings, drying and removal of the solvent, as
viscous dark purple goo was obtained, which smelled very strongly (and nicely) of raspberries. There again, it was impossible to obtain any
cristilline material from this substance ( mp pur rheosmin= 80°C).
The pink colour is a very curious thing: when collecting samples during the reaction, the pipet used to withdraw the supernatant turned pinkish after
a few minute sexposiiton to air. On the TLC plates, the saturated ketone stain turns dark pink whne exposed to UV! (and not before!) It gets darker
and darker when exposed several times... Quinonic material? Adding trace amounts of reductant makes the colour disappear, and when in solution in
AcOEt, it turns light red/orange....
I'm afraid the reaction conditions are not suited to phenolic unsaturated ketones, which must polymerise/dimerise much more easily than benzylacetones
and acetophenones... But the material does smell very nice!
So I'm going to work on other reductions, Cu/SiO2 seems to be the most promising, as CoCl2/NaBH4 requires 5x weight of the unsat ketone in CoCl2....
It's really a pity as this seme dto be the ideal reduction: very cheap Zn dust, easy reaction conditions and workup, etc Just too good to be true.. i
might try leaving the recation perform at room temp for 12h and see what happens.. Maybe dimerisation can be reduced enough to obtain a
cristallizeable product...
Also, this reduction could be applied to the ethers of phenolic unsaturated ketones: methylzingerone is said to smell of ginger too,
p-methoxyphenylbutanone is used in perfumery (not sure how it can smell though).
On the latest discoveries in the so-called Cassinone, 3,4-methylenedioxyphenylbutanone, a natural compound which is said to smell of blackcurrant, and
is used commecially. I think piperonal is fairly regulated in my country, so I will surely go via the benzyl halide and acetoacetate anion for this
one... That I will make in small quantities considering the lengthy preparation of the precursor.
Many thanks to Woelen for help with translations, and all the very kind people at the ref forums that helped me out with bibliographic research on
this subject...
Info on the Cu/SiO2 reduction will be summerized here, or a link to the Cu/SiO2 thread will be placed here.
Anyone with further suggestions on a easy, efficient reduction is very welcome to comment further..
[Edited on 23-9-2008 by Klute]
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
| Pages:
1
2
3
4 |
|








