| Pages:
1
..
7
8
9
10
11
..
19 |
aga
Forum Drunkard

Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
10ml α-pinene
20ml 15 v/v% H2SO4
125ml acetone
were mixed in a 250ml RBF and setup for reflux with stirring in an oil bath on a hotplate.
The mixture was held at between 85 and 90 C for 3.5 hours.
Boiling did not occur until 84 C, and the acetone was seen refluxing below the 1st bulb of the Allihn condenser at all times
(maybe the temperature can be raised to speed up the process).

After the 3.5 hours refluxing, the mixture remained a clear colourless liquid.

After briefly cooling in a water bath it became cloudy.

15 w% NaOH soloution was added in 1ml portions to the liquid with stirring to achieve a pH of 7~8. The amount added was 26ml.
The pH was tested by using half a pH strip on the end of tweezers to dip into the RBF.
Despite stirring, it was noted that there was a pH ~6 Upper layer in the liquid, as sampling required the pH strip to be immersed to a depth of at
least 3mm before the end of the strip showed any colour change.
After adding the NaOH, a large amount of white precipitate had formed, and the mixture had an oily upper layer.

The RBF was then setup for simple distillation to remove the bulk of the acetone, again in an oil bath on a hotplate with stirring.
(i find that stirring when distilling this kind of stuff eliminates bumping altogether)
After 25 minutes of distillation the vapour temperature began to climb, and the liquid in the boiling pot was showing phase separation, so the
distillation was stopped.
The residue in the RBF had become orange/brown in colour, with a small yet obvious oily upper layer.
The white precipitate had disappeared altogether.

The RBF was cooled in a water bath, then the contents transferred to a 100ml separatory funnel and left to stand for 5 minutes.

The lower aqueous layer was drained into a 100ml beaker.
After draining the lower layer, a very small third reddish layer was seen and drained off into a test tube.

Finally the upper (product) layer was drained into a 25ml RBF.
Total 'product' weight was 1.72g, although some of the red residue can be seen in the product.

[Edited on 26-12-2015 by aga]
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Very nicely executed, aga.
The "large amount of white precipitate" I suspect was Na2SO4, with the acetone acting as anti-solvent. When removing the acetone
that Na2SO4 then redissolved in the watery phase.
Mystery: the formation of reddish precipitate during the distilling off the acetone.
A bit disappointing: the small yield.
Encouraging: the formation of an organic layer during refluxing. It clearly shows some transformation of raw material.
Before anything else, any solids in the crude product will have to be removed before we attempt any further work-up. Perhaps filtration on a
mini-funnel with a high porosity filter medium?
For any follow-ups it would be wise to separate the organic and watery/acetone phases after the addition of the NaOH. I hadn't anticipated phase
separation at that point. (The acetone could then be recovered from the watery/acetone mixture.)
Ideas/suggestions for purification remain welcome. My idea was thermal crystallisation from a water/EtOH mixture.
[Edited on 26-12-2015 by blogfast25]
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
Quote: Originally posted by blogfast25  | | For any follow-ups it would be wise to separate the organic and watery/acetone phases after the addition of the NaOH. |
That did occur to me, just there was no room in the procedure for it.
The upper layer was quite large at that point - larger than the post-acetone-distillation upper layer.
I have a suspicion that the acetone removal might be avoided altogether with a little bit more 'salting out', perhaps with K2CO3
There are no solids i can see in the product.
The red stuff is also a liquid, although imiscible with the product
[Edited on 26-12-2015 by aga]
|
|
|
gdflp
Super Moderator
Posts: 1320
Registered: 14-2-2014
Location: NY, USA
Member Is Offline
Mood: Staring at code
|
|
Formation of a solid derivative, followed by recrystallization and decomposition would probably work quite well; especially on the small amounts which
aga has. Solid esters should work , as they allow for easy recovery of the alcohol through saponification. The only issue is that the required
reagents aren't necessarily the easiest to obtain, unless aga has some 3,5-dinitrobenzoyl chloride laying around. In this particular case however, the benzoate ester is almost certainly a solid as
well so benzoyl chloride could be substituted. Still not the easiest to obtain, but worst comes to worst, a straight fischer esterification with
benzoic acid would probably work instead at the cost of a slightly lower yield. In this particular case however, the benzoate ester is almost certainly a solid as
well so benzoyl chloride could be substituted. Still not the easiest to obtain, but worst comes to worst, a straight fischer esterification with
benzoic acid would probably work instead at the cost of a slightly lower yield.
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
Got some sodium benzoate if that helps.
Edit:
Might be a good idea to redo the whole thing and grab the stuff that separated after neutralising the acid.
[Edited on 26-12-2015 by aga]
|
|
|
gdflp
Super Moderator
Posts: 1320
Registered: 14-2-2014
Location: NY, USA
Member Is Offline
Mood: Staring at code
|
|
Great, conversion to benzoic acid is as easy as dissolving in water and acidifying with an acid of your choice.
Would you happen to have any S<sub>2</sub>Cl<sub>2</sub> still? If so, you can reflux it with benzoic acid to form benzoyl
chloride which can then be distilled off and used to directly react with the alcohol.
Remember, this is more involved then the crystallization from aq. EtOH, so I would call this more of a fallback idea if that doesn't work.
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
| Quote: Originally posted by gdflp | Great, conversion to benzoic acid is as easy as dissolving in water and acidifying with an acid of your choice.
Would you happen to have any S<sub>2</sub>Cl<sub>2</sub> still? |
Happily Not !
S<sub>2</sub>Cl<sub>2</sub> eats everything i put it in, including glass *
It can easily be made, so no difficulty in having some - briefly - if you have a reaction scheme in mind that needs it.
* it did not 'eat' glass, just that sulphur deposits stuck the stopper so hard that the rbf and glass stopper broke when i wanted to get it out.
[Edited on 26-12-2015 by aga]
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
Hold on a sec.
Would directly chlorinating the sodium benzoate not give NaCl and benzoyl chloride ?
|
|
|
gdflp
Super Moderator
Posts: 1320
Registered: 14-2-2014
Location: NY, USA
Member Is Offline
Mood: Staring at code
|
|
Unfortunately, no. There's an extra oxygen that you made disappear.
If you don't have any on hand, I don't think that the slight increase in yield is worth making it. As I said, I would try blogfast's idea first as
long as he has some sort of reference to back it up. I'm not sure how he wants you to crystallize it since it melts at ~ -36°C.
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
Ok.
We will have to await the Master's pronouncement then.
(Damn. I like making S2Cl2 - such an Interactive synthesis)
Meanwhile i think Plan B should be to grab that pile of stuff that floats after adding the acid neutralising agent.
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by aga |
Meanwhile i think Plan B should be to grab that pile of stuff that floats after adding the acid neutralising agent.
|
Totally Agreed. As I wrote: I didn't anticipate formation of an organic layer after neutralisation. It's a good sign, IMHO.
I'm not in favour of converting such a small amount to yet something else, for purification purposes only. It just adds more pieces to the puzzle.
So let me think about the next move, using what little we have as guinea pig.
Pure α-terpineol is reported to be a white, crystalline solid, MP 35 - 40 C. That's why I'm hoping some thermal re-crystallisation could
work. Need to think of the right solvent and quantities, though...
[Edited on 27-12-2015 by blogfast25]
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
From what I gather the -36 C is a Wiki error: most sources have the MP at +35 to +40 C. That makes more sense to me.
[Edited on 27-12-2015 by blogfast25]
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
Some crystals have formed in the 'product' overnight.

|
|
|
gdflp
Super Moderator
Posts: 1320
Registered: 14-2-2014
Location: NY, USA
Member Is Offline
Mood: Staring at code
|
|
| Quote: Originally posted by blogfast25 |
From what I gather the -36 C is a Wiki error: most sources have the MP at +35 to +40 C. That makes more sense to me.
|
Now that you say that, it does make a lot more sense. Considering that tertiary alcohols with fewer carbons, such as t-butanol, melt at ~25°C, it
follows that pure a-terpineol should melt at a higher temperature than that. I suppose I should have looked further than wiki.
Since that is the case, conversion to a benzoate is less useful, as the main purpose is to give a solid which can easily be recrystallized. The only
issue I can see with the thermal crystallization is oiling out if terpineol starts crashing out above 35°C.
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by gdflp | | The only issue I can see with the thermal crystallization is oiling out if terpineol starts crashing out above 35°C. |
The only other issue I see is that we know 'WTF' about solubilities of alpha-terpineol in various solvents. The only quantitative and
credible ref. so far is:
| Quote: | | 1:8 OR MORE IN 50% ALCOHOL |
https://pubchem.ncbi.nlm.nih.gov/compound/alpha-TERPINEOL#se...
One possibility could be cold acetone, then slowly adding cold water to the solution as an anti-solvent.
[Edited on 27-12-2015 by blogfast25]
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Wow. Didn't even see that there right away!
Fleshy computer now crunching away, meeting 'overload' errors...
Tidbit: I estimate the organic layer in the photo below (after reflux and after neutralisation) to be about 10 ml, from the other known dimensions in
the photo:
http://www.sciencemadness.org/talk/files.php?pid=432888&...
[Edited on 27-12-2015 by blogfast25]
|
|
|
gdflp
Super Moderator
Posts: 1320
Registered: 14-2-2014
Location: NY, USA
Member Is Offline
Mood: Staring at code
|
|
This lists the solubility in water as 2.42g/L @ 20°C, but it also lists the MP as -35°C so I'm inclined not to trust it. The most obvious thing
to try would just be a straight recrystallization from water or aq. EtOH; heating the solution up to 40°C, saturating with the crude terpineol, and
cooling down to 0°C to see if any crystals form.
|
|
|
gdflp
Super Moderator
Posts: 1320
Registered: 14-2-2014
Location: NY, USA
Member Is Offline
Mood: Staring at code
|
|
Just found data from the CRC(2003), which lists the properties as follows :
MP : 40.5°C
BP : 220°C
Solubility: sl H<sub>2</sub>O; vs acetone, benzene, ether, EtOH
Crystalline Form : Crystals, Petroleum Ether
It might be worth a try then recrystallizing from pet. ether as well.
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by gdflp | | This lists the solubility in water as 2.42g/L @ 20°C, but it also lists the MP as -35°C so I'm inclined not to trust it. The most obvious thing
to try would just be a straight recrystallization from water or aq. EtOH; heating the solution up to 40°C, saturating with the crude terpineol, and
cooling down to 0°C to see if any crystals form. |
If it crystallises out of it's own impurities, as suggested by aga's latest post, then that might be enough: remember that this isn't the end-product,
we need to hydrogenate the alpha-terpineol.
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
It has occurred to me but I don't think aga has any Pet. ether. Hexane or cyclohexane would probably work too. Or 'octane': distilling lead-free
petrol is fun!
Thanks for the ref.!
[Edited on 27-12-2015 by blogfast25]
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
I only have furry Pets.
Petrol distillation ? OK !
I'll use the Bigger burner.
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Oh and here's the proposed reaction mechanism for the conversion of α-pinene to α-terpineol:
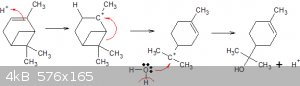
1. A proton/oxonium electrophilic attacks the double bond, creates carbonium ion.
2. Orbital movement causes the charge to move to the tertiary carbon atom (more stable), creates new double bond.
3. Nucleophilic attack by water on that carbonium atom creates desired carbinol and releases proton (confirming protons as catalyst).
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Hold yer fire and put yer hands above yer heads!
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
lol
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
OK.
Boiling off the acetone appears to have knackered the product, or at least reduced yield to almost nothing.
I think the consensus is that 'we don't know' so time for some experimentation, surely ?
Firstly a Bigger batch needs making, say 2x or 3x the original volumes.
From that there should be around 20~30ml of raw product as per blogfast's calculation from the photo.
Let's chop the post-reflux liquid into 4 equal portions and do different things to it to see what happens.
As there was phase sep and some crystals without even trying, i suspect that neutralising the acid with an excess of K2CO3 will
be enough to get the product out.
That's 1 portion taken, just need 3 other suggestions for the others ...
As the acetone was barely even reaching the first bulb in the condenser during reflux, could the heat be turned up to reduce reaction time (e.g.120 C)
or would excess heat lead to other non-desireable products ?
Edit:
In the reaction scheme, the acetone is absent.
If it is merely a solvent, then the sheer quantity specified is vastly greater than it needs to be : during reaction the drip rate from the condenser
is around 2 drops a second or less @ 85 C.
[Edited on 27-12-2015 by aga]
|
|
|
| Pages:
1
..
7
8
9
10
11
..
19 |








