Boffis
International Hazard

Posts: 1852
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Mono-nitration of resorcinol
For no particular reason a few days ago I decided to try the mononitration of resorcinol. Knowing that phenol tends suffer oxidation during
mononitration; if sulphonated first the tendency to suffer oxidation is reduced but you end up with either polynitrophenol or nitrophenol sulphonic
acids. Phenol can be nitrate with fairly dilute nitric acid alone (see http://www.sciencemadness.org/talk/viewthread.php?tid=155527...)
With this in mind I decided to try a similar method of nitration as but because resorcinol is less soluble I tried to dissolve it in 70% sulphuric
acid first. This was not particularly successful and was going to require a lot of acid so I added an equal volume of conc. sulphuric acid and warmed
it gently until complete solution occurred and then chilled it in the freezer to minimise sulphonation. A small excess of 50% nitric acid was then
added the cooled mixture dropwise. The reaction solution rapidly turned red and after standing at room temperature for an hour or so had turned a very
intense maroon red. The product recovered from this solution is a deep red crystalline solid, slightly soluble to give a deep red solution in cold
water and freely soluble in hot water, less soluble in alcohol and IsoPA even when hot. It gives a brownish purple colour with FeCl3 solution and a
brown colour with dilute NaOH.
Experimental details
5.010g of resorcinol were dissolved 7.5ml of 70% and 7.5ml of 96% sulphuric acid with gentle warming and then the solution chilled to about 0 C in the
freezer. 3ml of 50% nitric acid were added dropwise to the solution which soon turned red and after an hour had turned intense maroon red. The
reaction mixture was poured into 30ml of ice cold water and the solids filtered off. The filtrate was neutralised with 40% NaOH solution but deposited
only much sodium sulphate in colourless crystals superficially stained red and was discarded.
The solid was dried and then an attempt made to recrystallise it from iso-propanol but it proved to be very sparingly soluble in this solvent even
when boiling. It could be recrystallised from water, about 10ml where required. On drying thin, colourless blades formed by efflorescence on the
surface, the red solid consisted of minute red resinous spheres.
The question is; what is it, the blades are probably unreacted resorcinol but the rest? What type of compound is it? The deep red colour suggests an
azo compound so what sort? A poly hydroxy-azobenzene? Perhaps:

|
|
|
Fery
International Hazard
Posts: 1009
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
Hi Boffis. I nitrated phenol and I would never repeat it again, it was the most messy experiment I did in my life. Luckily I cleaned everything with
NaOH solution, even traces of nitrophenols made everything yellow.
Solubility of resorcinol in water should be 110 g per 100 ml at 20 C but certainly less at lower temperatures.
https://en.wikipedia.org/wiki/Resorcinol
So couldn't be resorcinol just dissolved in water and then HNO3 added when cooling in ice bath to e.g. 0 C? I nitrated phenol with 30% HNO3 so
resorcinol is even more reactive.
https://www.sciencemadness.org/whisper/viewthread.php?tid=15...
From literature about phenol nitration the treatment with NaOH is discouraged, it allegedly produces some polymers or something not desirable.
Deep colors are perhaps oxydation products of resorcinol and later due to addition of NaOH. Even invisible traces of nitrophenol turned into
intensively yellow when washing glass with NaOH.
You can perhaps produce the compound on the picture from nitroresorcinol when reacting with Mg in methanol. I did similar reduction and got azobenzene
from nitrobenzene. But that would require extremely dry methanol, even traces of water badly delay the start of reaction (I used methanol where label
stated at most 0,05% water, but better would be to use methanol with not more than 0,01% water).
https://www.sciencemadness.org/whisper/viewthread.php?tid=15...
|
|
|
Fery
International Hazard
Posts: 1009
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
here I found useful interesting experiment they nitrated various substituted phenols:
https://sci-hub.ee/10.1021/op0200120
Attachment: op0200120.pdf (95kB)
This file has been downloaded 252 times
they used phase transfer catalyst TBAB tert butyl ammonium bromide, very diluted HNO3 (6%), organic layer dichlorethane (maybe to immediately capture
mononitrophenol to prevent its oxidation or dinitration ?) and got nice yields like over 80% (when I nitrated phenol I got only 50%)
| Quote: | 4-chlorophenol (10
mmol), TBAB, 10 mol % of substrate, nitric acid (70 wt %,
20 mmol), water to make up the nitric acid concentration to
6 wt %, and ethylenedichloride (20 mL), were mixed together
in a 50-mL glass reactor and stirred at 20 °C for 4-6 h.
Reaction progress was monitored by GC. Products were
identified by GC-MS, and the data were compared to those
of the authentic samples purchased from commercial sources.
After the completion of reaction, the organic layer was
separated from the aqueous layer. It was then washed with
3 × 25 mL water, separated, and finally dried over
magnesium sulfate. The anhydrous ethylenedichloride layer
thus obtained was distilled to remove solvent. The residue
was then crystallized in ethanol to obtain 1.41 g (81 mol %)
of 4-chloro-2-nitrophenol. The observed mp was 86.5 °C
which was in good agreement with the literature (mp 85-
87 °C). |
|
|
|
Lionel Spanner
Hazard to Others
Posts: 166
Registered: 14-12-2021
Location: near Barnsley, UK
Member Is Offline
|
|
I suspect you have a mixture of nitroresorcinols, some of which are sulphonated - which would explain why they're readily soluble in water.
Nitroanilines absorb in the visible spectrum, often with an intense red or orange colour (and are in fact used as dyes), so it's reasonable to suppose
that nitroresorcinols would have a similar colour.
The brown colour from sodium hydroxide is most likely from polymerisation. From my experience in formulating oxidative hair dyes, resorcinol
derivatives tend to produce brown colours when undergoing oxidative polymerisation in basic conditions.
|
|
|
Fery
International Hazard
Posts: 1009
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
Resorcinol 22,0 g (0,2 mol, Molar mass 110.1 g/mol) dissolved in 22,0 g of water - yes I weighed it, I did not measure volumes too much (seems to be
endothermic, it was necessary to warm to 25 C to dissolve and then cool).
0,2 mol HNO3 = 12,6 g 100% = 22,1 g 57% (Molar mass 63.01 g/mol) (I used 22,8 g of 57% HNO3 which is small excess, there are small wastes of acid
adhering on walls of dropping funnel, excess of acid makes final product even less soluble than in neutral aqueous solution due to supression Ph-OH +
H2O -> Ph-O- + H3O+)
Resorcinol solution cooled in water-snow bath, when T of solution decreased to 10 C (no crystallization of resorcinol observed) dropping HNO3
commenced at a rate that the T of reaction was kept at 10 C. Soon turbidity observed at which I was unable to distinguish whether crystals or droplets
of immiscible liquid (mononitroresorcinol ???).
HNO3 dropping finished in 1 hour in cold bath, T of reaction 10 C. Then stirred for further 3 hours during which T allowed to slowly raise to 20 C.
No red fumes of nitrogen oxides observed.
Cooled outside to -5 C (winter temperature), sucked on cold sinter outside (-5 C).
Wet product 23,1 g
Mother liquor 40,7 g
0,30 g NaOH (7,5 mmol) dissolved in 2 ml water and the mother liquor added in small portions, in the beginning the color yellow in alkaline
environment, when changed to acidic the color the same as mother liquor (just a little paler) - red.
Totally 5,2 ml of mother liquor added to NaOH to observe again red color. I did not titrate, it was just random addition of small portions. So perhaps
slightly less volume would neutralize the hydroxide.
So 5 ml of mother liquor has 7,5 mmol HNO3 (M=63 g/mol) = 0,5 g HNO3 which is still 10% HNO3
there is 8 x more of red liquid = 4,0 g HNO3 did not react
22,8*0,57 = 13,0 g HNO3 was added to reaction, 9,0 g HNO3 reacted (70%) and 4,0 g didn't (30%).
Maybe excess of HNO3 should be used, in the attached experiment in my previous post they used 2-fold excess (I used stoichiometric amounts to just
study the reaction and to certainly avoid dinitroresorcinol formation)
reactants 22+22+23=67g
products 23+41=64 g
3 g lost on reaction flask walls and filtration apparatus, especially porous sintered glass, porosity 4)
from fenol nitration I got 50% yield
I can't process the wet product further yet, I'll be back in lab on new year. According the above approximate calculations I hope the product is
mostly 4-nitroresorcinol and not only reactant crystallized on cooling down.
 Sitting already in a train. Who on Earth invented Christmas??? Sitting already in a train. Who on Earth invented Christmas???
   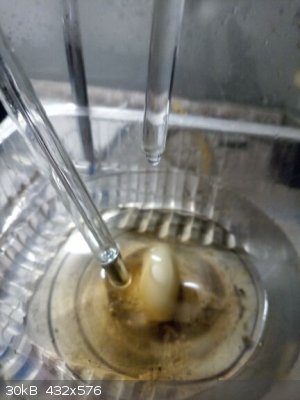 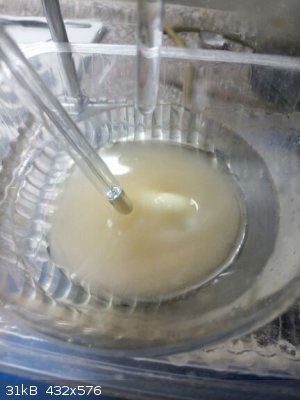    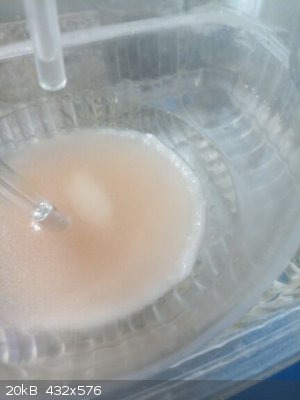     
|
|
|
clearly_not_atara
International Hazard
Posts: 2754
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
https://pubs.rsc.org/en/content/articlelanding/1958/jr/jr958...
|
|
|
Boffis
International Hazard
Posts: 1852
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
@Fery; thanks for the lead on this nitration technique. I'll give it a go on resorcinol. But this still doesn't explain what the red material is.
Incidentally, I found several preparation for mono nitro resorcinols in the old literature and they are all heavily convoluted, requiring either oleum
sulphonation of the resorcinol first then boiling with acid to remove excess sulphonic acid groups etc but all of the nitro compounds are described as
yellow crystals.
So Lionel Spanner I don't the red product is a simple nitro resorcinol or their sulphonic acid derivatives. As I said the compound resembles some of
the hydroxy azobenzene type compounds. Azoxy compounds tend to be less intensely coloured. I have just dug out some Na dithionite and I will try
reducing it tomorrow.
Sorry Fery, I hadn't seen your nice preparative post when I made my post, that is an interesting and informative post. I find it curious that quiet a
few simple phenolic mononitro compounds seem poorly described in the literature in spite of them potentially very useful intermediates.
C_n_A; That's an interesting paper. You wouldn't think that there would be much nitrite in my original system but it is possible since the nitric acid
I used was diluted from some very old high-test nitric acid (originally 85-88%) and was rather yellow. So maybe that's the answer.
[Edited on 23-12-2021 by Boffis]
|
|
|
Fery
International Hazard
Posts: 1009
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
The sulfonation prior nitration seems to be necessary for preparing 2-nitroresorcinol.
https://sci-hub.ee/10.1021/ed047p224
The 2-derivate is described as red-orange and distills with steam the same way as 2-nitrophenol (which is yellow). 4-nitrophenol does not distill with steam. I do not know how it is with 4-nitroresorcinol according steam distillation. It is similar to
2-nitrophenol as the hydroxyl in position 3 creates H...O bridge with nitrogroup at position 4, but the hydroxyl in position 1 is so faraway from
nitro group as the positions in 4-nitrophenol.
melting points:
https://en.wikipedia.org/wiki/Resorcinol
resorcinol
Melting point 110 °C
https://www.chemicalbook.com/ChemicalProductProperty_EN_CB59...
4-nitroresorcinol
m.p. 122 C
https://www.fishersci.com/shop/products/2-nitroresorcinol-tc...
2-Nitroresorcinol
Melting Point 83 °C
|
|
|
|










