| Pages:
1
2 |
Magpie
lab constructor

Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
Preparation of Salicylaldehyde
a lab report
A. Introduction
Salicylaldehyde (SA) can be prepared using a Reimer-Tiemann reaction, ie:
Attachment: Reimer-Tiemann.doc (26kB)
This file has been downloaded 833 times
reaction
This reaction proceeds through formation of a :CCl2 carbene as shown in reference 1.
I followed the procedure found in Vogel’s 3rd ed, page 703 (ref 2). The expected yield is 12g. My crude flowsheet for this preparation is shown
below:
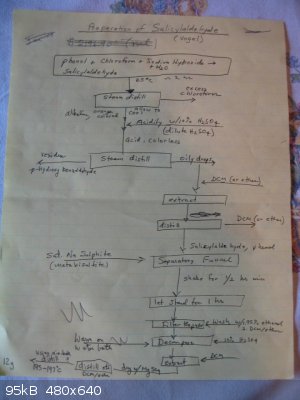
SA preparation flowsheet
B. Apparatus
3-neck 1000 ml RBF (24/40)
overhead stirrer
west condenser (24/40)
west condenser (19/22)
60 ml p-e addition funnel
hotplate
thermocouple w/readout
2-quart waterbath (steel bowl)
distillation head (24/40)
distillation head (19/22)
vacuum adapter (19/22)
adapter (24/40 to 19/22)
100 ml RBF (19/22)
25 ml RBF (19/22)
cooling water (iced) supply
1000 ml heating mantle
100 ml heating mantle
aluminum foil
fiberglass insulation blanket
thermometer good to 200°C
steam distillation apparatus:
1. 500 ml separation funnel (steam trap)
2. 5mm x 30cm glass tube
3. 2-hole rubber stopper
4. ¼” latex rubber tubing, ~2’
5. hot plate
6. pressure cooker
7. thermometer adapter (19/22)
8. adapter (24/40 to 19/22)
9. 2ea glass plugs (24/40)
125 ml separatory funnel
250 ml separatory funnel
miscelaneous beakers
C. Reagents
28.4g phenol (aqueous, 88%)
40.5 ml chloroform
80g NaOH
water
1M H2SO4 (~500 ml)
dichloromethane (~ 85 ml)
anhydrous MgSO4
sodium bisulfite (or sodium metabisulfite)
D. Procedure
1. Reaction
The reaction apparatus was set up as shown below. The 2nd picture shows the utilities outside the hood that were required for support.

SA reaction setup

SA utilities – outside the hood
To the 1000ml RBF was added a warm solution of 80g NaOH dissolved in 80 ml of water. To that was added a solution of 28.4g of 88% phenol dissolved in
21.6 ml of water. The water bath was kept at about 65°C. 40.5 ml of chloroform was added to the 60ml p-e funnel attached to the outlet of the
condenser using a notched rubber stopper.
40.5 ml of chloroform was added to the p-e funnel. With the overhead stirrer running at 222 rpm the chloroform was added slowly over a period of ½
hour. I attempted to keep the temperature of the reactants at 65-70°C per procedure by adding boiling water to the water bath. However, the
temperature ranged from 59°C to 64°C. I didn’t turn on the hotplate as I was afraid of a possible runaway. This never happened and I never did
have to cool the bath. The mix was then heated with the water bath boiling for another hour.
During the addition of the chloroform the reactant mix turned red and eventually turned to a dark brown, looking very much like beef gravy.
Chloroform refluxed all the while. There was foaming for a time but it was not a problem due to the generous freeboard of the 1000ml RBF. At cleanout
of the RBF a disc of tar about the size of a 50 cent piece was found at the bottom of the flask. It was soluble in alcohol and a tiny bit gave a
strong yellow coloration - dare I say à la Perkin. Pictures of the reactants at various stages are shown below:

SA reactants – later – after all chloroform added

SA reaction – during final 1 hour hold
2. Workup
The workup for this synthesis is very long, nearly discouraging me from even starting this synthesis. Garage chemist described it succintly as “a
pain.” See the flowsheet above.
a. Steam distillation to remove unused chloroform
This was performed in the 1000ml pot as shown below. I recovered no chloroform. I believe that any unused chloroform escaped from the 24/40 West
condenser (on ice-water). Vogel calls for “an efficient (double wall) reflux condenser.” I don’t have one of those. The picture below shows
the steam distillation.

Steam distillation to remove excess chloroform
b. Acidification
The pot mix was then acidified with ~250 ml of 1M H2SO4. The color of the mix lightens considerably during the acid addition.
c. Steam distillation to remove the SA/unreacted phenol
A second steam distillation was now done to remove the SA and any unreacted phenol as shown below:

2nd steam distillation to remove SA/phenol
d. Extraction
The distillate from the 2nd steam distillation was now extracted with 25ml x 25ml dichloromethane (DCM) to remove the SA/phenol from water. This is
shown below. The yellow liquid in the beaker is the first extraction, the clear liquid in the bottom of the funnel is the second extraction.

SA/phenol extraction with DCM
e. Distillation
The combined extracts are distilled to remove DCM. 45ml of good quality DCM was obtained for recycle. This is done using a water bath as shown
below:

Removal of DCM by distillation
This report is continued on the next post due to the excessive number of images.
[Edited on 30-3-2015 by Magpie]
[Edited on 30-3-2015 by Magpie]
[Edited on 31-3-2015 by Magpie]
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
morganbw
National Hazard
Posts: 561
Registered: 23-11-2014
Member Is Offline
Mood: No Mood
|
|
Looking forward for the workup.
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
Continuation
f. Sulfite adduct formation
The sulfite adduct is now formed to separate the salicylaldehyde from the excess phenol. This is done by adding 2X its volume of saturated aqueous
sodium bisulfite (I used Na metabisulfirte). Since the mix is biphasic Vogel called for this to be shaken vigorously for 1/2hr, then allowed to sit
for 1 hour. The resting product is shown below:
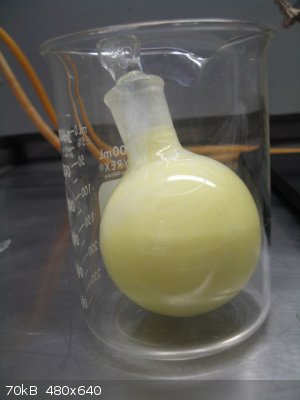
SA bisulfite adduct – post shaking
g. Separation of the adduct by filtration
The adduct, a pure white solid, was then separated from the water and phenol by filtration using a 7cm Buchner filter. It is shown in the picture
below:

SA adduct filtered
At one point during the filtration I did not get the filter paper properly sucked down (sealed) against the ceramic base. Consequently I lost some
product here – possibly as much as a gram. The dried adduct was weighed (17g), indicating the presence of 9.2g of SA.
h. Recovery of the salicylaldehyde
The SA is recovered by decomposition of the adduct. This is done by adding 1M H2SO4 then gently warming in a water bath. I added 75ml of the acid.
During the warming I got too aggressive with the heating in an effort to fully decompose the adduct to SA and SO2, forgetting to follow the direction
of “warming.” As a consequence I believe I lost 2-3g of product via steam distillation up the hood vent. I could visibly see that the volume of
yellow oil on the bottom of the flask had decreased. Warming the adduct in the water bath is shown below:

SA bisulfite adduct decomposition
i. Extraction
The SA/water product was then extracted with 25ml x 15ml of DCM. The extract is shown drying overnight over anhydrous MgSO4 in the picture below:

SA in DCM being dryed
i. Distillation
The next day the DCM was removed by distillation. Following this with the pot heavily insulated with a fiberglass blanket shrouded in aluminum foil,
the SA was distilled. The product was captured that boiled in the range of 193-196°C. Literature value (Vogel) is 195-197°C. Pictures of the
distillation apparatus, the thermometer at product take-off, and the salicyaldehyde product are shown below:

SA distillation

SA distillation temperature

SA product
E. Results
My yield was a disappointing 6g. The yield would have been better if not for the losses described above. The expected yield (Vogel) is 12g. Even
this is only a 37% yield.
F. Conclusions
1. Salicylaldehyde can be successfully made using the Riemer-Tiemann reaction and Vogel’s procedure. My poor yield can certainly be much improved.
2. DCM can be successfully substituted for diethyl ether as the extractant in this synthesis. Other than its obvious advantages I think DCM gives
cleaner separations, and is quite recoverable by distillation.
3. My homemade overhead stirrer performed satisfactorily.
G. Discussion
My reasons for doing this synthesis are many: (1) I wanted to perform a complex synthesis involving relatively benign chemicals as I have been out
of the lab for ~3 months due to a health issue which has since been satisfactorily resolved, (2) I had 500ml of reagent grade phenol that I’ve had
for over 5 years without use, (3) I wanted to find out how well DCM could be substituted for diethyl ether for extraction, (4) I wanted to test out my
new stepper motor overhead mixer, and (5) JimmyMajesty’s work with aldehydes piqued my interest in isolating an aldehyde by sulfite adduct. Also I
wanted to perform a name reaction new to me. I found the Riemer-Tiemann mechanism to be fascinating; and carbenes are always fascinating.
Salicylaldehyde has a smell very similar to benzaldehyde, but with a little bite and is not as sweet or strong. One source said it smells of bitter
almond.
H. References
1. http://en.wikipedia.org/wiki/Reimer%E2%80%93Tiemann_reaction
2. Practical Organic Chemistry, 3rd edition, by Arthur I. Vogel, 1956 (see forum library)
Questions, comments, and suggestions are welcomed.
[Edited on 30-3-2015 by Magpie]
[Edited on 30-3-2015 by Magpie]
[Edited on 31-3-2015 by Magpie]
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
That looks very interesting, Magpie, but I think I'll pass on trying it myself. 
|
|
|
morganbw
National Hazard
Posts: 561
Registered: 23-11-2014
Member Is Offline
Mood: No Mood
|
|
Good job, it would take many of us more than one try, to get any product.
|
|
|
Loptr
International Hazard
Posts: 1348
Registered: 20-5-2014
Location: USA
Member Is Offline
Mood: Grateful
|
|
You are on of my inspirations in this hobby!
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
Thank you blogfast, morganbw and Loptr.
I hope JimmyMajesty tries isolating his acetaldehyde by sulfite adduct.
[Edited on 31-3-2015 by Magpie]
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
Crowfjord
Hazard to Others
Posts: 390
Registered: 20-1-2013
Location: Pacific Northwest
Member Is Offline
Mood: Ever so slowly crystallizing...
|
|
Did you only decompose the solid adduct? A fair amount tends to remain dissolved in the aqueous filtrate (see Len1's Illustrated Practical Guide for
benzyl chloride, benzal chloride, benzaldehyde, etc in prepublications). Could have been another contributor to your low yield. I'm away from my
notes, but in isolating 2,5-dimethoxy benzaldehyde from reaction solvent and starting compound (Beauveault aldehyde synthesis) via bisulfite adduct, I
think I actually got more aldehyde from the aqueous solution than the filtered solid.
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
No, I did not. The adduct that slipped under the filter paper would have been in there too, along with the phenol, and a little alcohol/DCM wash. I
should have reviewed Len1's benzaldehyde synthesis.
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
Waffles SS
Fighter
Posts: 998
Registered: 7-12-2009
Member Is Offline
|
|
That is Great,
Magpie,i want to suggest another method
As you know Riemer-Tiemann is low yield method but o-formylation of phenols by mg has better yield.
Have you ever tried Mg-mediated o-formylation of phenols method?
also there is another interesting method according to Mg-mediated o-formylation of phenols.
I want to try it and will share the result:
| Quote: |
The One Pot Synthesis Salicylaldehyde Prepared by Reactive Grinding with Derivative Phenol and Paraformaldehyde in Absence Magnesium
Methoxide
A aromatic Phenol (0.1 mol), magnesium methoxide (5 gr ), and the mixture was grinding to 1 minute at room temperature. A slurry of paraformaldehyde
powder (4 gr) in under grinding was added in small portions over 2 min to the reaction mixture. Stirring was continued at r.t for 8 min, after which
added slowly to 10% sulfuric acid (20 ml g). The resulting mixture was stirred for 10 min, after which the aqueous layer was separated and extracted
with ethyl acetate (2 x 100 ml). The combined organic layers and extracts were washed with 10% sulfuric acid (20 ml) and water (20 ml) and evaporated
under reduced pressure to give the salicyaldehyde (Yield 85%)
http://dx.doi.org/10.13005/ojc/290445
|
and also below:
| Quote: |
Magnesium-mediated ortho-Specific Formylation and Formaldoximation of Phenols
2-Hydroxybenzaldehyde -Phenol (37.6 g, 0.4 mol) was
added to magnesium methoxide (259 g of 8 wt.% solution in
methanol; 20.7 g, 0.24 mol) and the mixture was heated to
reflux. Approximately half the methanol was distilled off and
toluene (300 g) was added to the residue. The azeotropic
mixture of toluene and methanol was removed by fractional
distillation, until the temperature of the reaction mixture rose to
95 "C. A slurry of paraformaldehyde powder (43.2 g, 1.44 mol)
in toluene (75 g) was added in small portions over 1 h to the
reaction mixture at 95 "C with concurrent removal of volatile
materials by distillation. Stirring was continued at 95 "C for 1 h,
after which mixture was cooled to 25 "C and added slowly to
10% sulfuric acid (450 g). The resulting mixture was stirred at
30-40 "C for 2 h, after which the aqueous layer was separated
and extracted with toluene (2 x 100 g). The combined organic
layers and extracts were washed with 10% sulfuric acid (50 g)
and water (50 g) and evaporated under reduced pressure to give the aldehyde 2a as a pale yllow oil (48.35 g, 84%)
Aldred, Robert; Johnston, Robert; Levin, Daniel; Neilan, James; Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic
Chemistry (1972-1999); nb. 13; (1994); p.1823 - 1832
DOI: 10.1039/P19940001823
|
[Edited on 31-3-2015 by Waffles SS]
|
|
|
Loptr
International Hazard
Posts: 1348
Registered: 20-5-2014
Location: USA
Member Is Offline
Mood: Grateful
|
|
How sensitive are aldehydes to air oxidation? Any particular measures that must be taken during the procedure, or doing its storage? I know you
previously synthesized benzaldehyde using persulfate, and am curious if the oxidation potential is the same for both salicylaldehyde and benzaldehyde.
|
|
|
lullu
Hazard to Self
Posts: 51
Registered: 2-3-2012
Member Is Offline
Mood: No Mood
|
|
Very nice Magpie!
Is there any specific reason you liberated the aldehyde in acidic conditions, thereby forming SO2?
Or just because you can as you have one of the nicest fume hoods I've seen on here  ? ?
I would also recommend the Mg formylation as an alternative it is a breeze.
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
Thank you waffles and lullu. I used the Reimer-Tiemann for the reasons stated in the "Discussion" section of my report. But thanks for the
information on o-formylation.
lullu, that is an interesting question about using acid for liberation of the aldehyde from the adduct. I've seen for other syntheses, and expected,
that this would be done with a base. I just followed the direction in Vogel in using the acid, 1M H2SO4.
Loptr, I can't say for sure for salicylaldehyde (yet) but for benzaldehyde it is surprisingly stable. I would expect SA to be similar. But for
butyraldehyde and propionaldehyde my experience is that they oxidize easily.
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
Waffles SS
Fighter
Posts: 998
Registered: 7-12-2009
Member Is Offline
|
|
Are you sure Benzaldehyde is stable in oxidation?
According to my experience it will oxidized almost fast with air oxygen .
Also it is suggested by different resource to distill it before use or wash it with Carbonate solution for get rid of Benzoic acid
Oxidation of Salicyldehyde depend on oxidation substance.
Usually Electron donating group(activating groups) make Benzaldehyde more stable in radical oxidation but for OH group i think we should test it.
[Edited on 31-3-2015 by Waffles SS]
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
I made my benzaldehyde some time ago and have used it at least 2 times since for successful syntheses. That's all I have to go on.
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
NexusDNA
Hazard to Others
Posts: 104
Registered: 23-11-2013
Location: Brazil, under an umbrella
Member Is Offline
Mood: Liberated from cocoon
|
|
Excellent, Magpie! Thank you!
I'm interested in doing this preparation. I'll post here if everything goes ok.
Bromine, definitely bromine.
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
Thank you Nexus.
After recovering the 2nd batch of DCM (40 mls) I gave it a sniff test (not recommended) for quality control before placing it in the mother supply
bottle. It had a sharp, nasty sting so I set it aside for later cleanup w/activated charcoal. After running it through the charcoal I checked again
with a sniff (not recommended) and - ouch - it had not changed. But this time I realized what it was: SO2!
Of course - it had been dissolved during the extraction following the adduct decomposition. So, I treated it with an aqueous CaCO3 slurry. This was
a mistake as the CaCO3 seemed to react with the DCM forming a gummy mess. It did get rid of the SO2 however, so I did recover about 20 mls. Na2CO3,
NaOH, or KOH, would probably have been a good choice.
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
Jimmymajesty
Hazard to Others
Posts: 153
Registered: 9-7-2009
Member Is Offline
Mood: No Mood
|
|
Hey Magpie,
Well done!
I tried to isolate the acetaldehyde as the bisulhite adduct.
It needs time for the formation and for the decomp also, also you can precipitate the bisulphite without the adduct if there is an ethanol carry over
and there is always some with the catalytic method. So I do not isolate the acetaldehyde rather use it as a gas, but if you need some more info on the
acetaldehyde adduct I surely can do some experiments for you
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
Thanks Jimmy. No, I have no plans to make acetaldehyde at this time. But I might in the future, as like I said before, for some reason I want to
make crotonaldehyde. I just wanted you to see how the adduct is used in case you wanted to use it to isolate your acetaldehyde or store it in powder
form.
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
learningChem
Hazard to Others
Posts: 182
Registered: 21-7-2011
Member Is Offline
Mood: No Mood
|
|
Would the reaction work with cresol ? (yielding some kind of methyl aldehyde?)
|
|
|
CaptainPike
Hazard to Self
Posts: 68
Registered: 21-12-2012
Member Is Offline
Mood: No Mood
|
|
Magpie,
We love you – you make the absolute best OC Porn, ever!
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
Thanks, Captain.
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
ronstark
Harmless
Posts: 37
Registered: 10-10-2016
Member Is Offline
Mood: No Mood
|
|
Did try the adduct for the first time to improve my chemistry. It was made according to Vogel's: 50g Sodium Bisulfite in 100mL water and 70mL EtOH.
Some chunks remained undissolved, but filtered to a clear solution. 50g Salicylaldehyde added to the solution and stirred 1h. The crystals were
filtered and the filtrate had a blue purplish colour. Is this normal? After the filtrate was dried, 90g SA adduct. 100mL 10% NaOH was added and
crystals begin to form on the bottom. The solutiom is reddish and on top oily drops, more like chicken fat. Can I saturate this with NaCl? Think I
will try with H2SO4 next time. Looks more clean on your photos. I did extract a portion with DCM and upon evaporation it was a red purple oil. It
seems very hard to extract that oily drops from the solution. Xylene is a better solvent?
[Edited on 9-2-2017 by ronstark]
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
I saw no colors after the formation of the adduct other than a slight yellowing of the final product.
I have no special insight into where your blue and red colors came from. My only guess would be impure product/reagents somewhere along the line.
There could be a reason for Vogel's specification of 1M H2SO4 instead of using NaOH for regeneration of the aldehyde.
I don't know if xylene would be a suitable solvent as it is completely non-polar. DCM and ether have some polarity, however, which would seem more
appropriate.
[Edited on 10-2-2017 by Magpie]
[Edited on 10-2-2017 by Magpie]
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
UC235
National Hazard
Posts: 565
Registered: 28-12-2014
Member Is Offline
Mood: No Mood
|
|
Purplish color sounds like iron contamination to me with salicylic acid around.
I looked into the Reimer-Tiemann reaction a while ago for salicylaldehyde since I lack paraformaldehyde, but except for certain substrates, the yield
is quite terrible. The magnesium mediated approach is vastly superior.
Attachment: ReimerTiemannreview.pdf (1.4MB)
This file has been downloaded 1578 times
|
|
|
| Pages:
1
2 |









