nimgoldman
Hazard to Others

Posts: 303
Registered: 11-6-2018
Member Is Offline
|
|
Unable to concentrate nitric acid to 68%
I usually make concentrated nitric acid by the same simple procedure as presented many times over, like here, except I use 30 cm Vigreux column for the second distillation.
I decided to actually measure the density and to my surprise, it was only 1.34 g/mL ... this means my acid is only 58%, not 68%. This baffles me since
people are easily able to achieve azeotropic nitric acid with second simple distillation and the fractional should perform even better, right?
My crude nitric acid (before fractionation) was 53%, the solution started boiling at 115 °C (239 °F) and the temperature at the still head was 110
°C (230 °F) instead of expected 100 °C (212 °F). So instead of removing mostly water, lots of acid has been removed as well.
Then I wait until the head temperature raises to about 118 °C (244.4 °F) and switch the flasks. If I wait longer, a good half of the volume in the
boiling flask gets over before the head temperature finally reaches 120 °C (248 °F) which is almost the b.p. of nitric acid azeotrope. The fraction
taken above 118 °C (244.4 °F) has a concentration of 58%, not much close to 68%.
I don't understand why the Vigreux column provided worse fractionation than a simple distillation people often use to make 68% nitric acid?
I used aluminium foil insulation on the column but maybe it is not beneficial in this case? But even if the column was perfectly insulated, it would
still at worst perform like a simple distillation, which is known to provide azeotropic or near-azeotropic nitric acid.
Should longer column fix the problem?
Since my fume hood is too small for an entire distillation apparatus, I use a hose attached to the vacuum adapter to take the nitrogen dioxide into
the fume hood and then outside. Maybe just having the long hoe attached raises the pressure in the apparatus enough to spoil fractionation? I think
this is improbable but I am out of ideas...
I will try titration to measure concentration more precisely, sine maybe I made a mistake in the density measurement (I use volumetric flask and a
scale).
Or maybe the temperature measurements were off due to a different atmospheric pressure in my area (474 meters above sea level) - for water, this
should lower the b.p. of water by almost 2 degrees. But this does not explain why the first fraction comes over actually at higher temp, not lower.
|
|
|
Sulaiman
International Hazard
Posts: 3559
Registered: 8-2-2015
Location: 3rd rock from the sun
Member Is Online
|
|
First, it would be simpler to initially add only the quantity of water requied to produce (slightly greater than) azeotropic concentration in the
original reaction,
this way the distillation is not actually required, just removal of the sulphate salt (and other impurities) by cooling and filtration,
A simple distillation would of course give a purer product.
As (drain unblocker) sulphuric acid is relatively cheap,
it can be used to de-hydrate dilute nitric acid to any concentration prior to distillation
===============================================
Here is a diagram of nitic acid vapour and liquid concentrations vs temperature
( taken from https://www.chemguide.co.uk/physical/phaseeqia/nonideal.html )
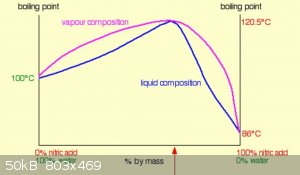
Looking at that diagram you can estimate that two 'theoretical distillation plates' should concentrate 53% to very nearly azeotropic,
the boiling pot/rbf is one 'plate' and a Vigreux column should easily provide the rest.
So in theory, from your 53%, you should have obtained near azeotropic nitric acid.
Nitric acid concentration determination by density is VERY temperature sensitive,
did you compensate for temperature:density ?
......................................................................................................
P.S. many nitrations can be performed using in-situ generated nitric acid
(i.e. using sulphuric acid and a nitrate salt instead of nitric acid)
which saves time/effort.
I like to use excess sulphuric acid and no additional water in the starting reaction,
on distillation this gives RFNA and some waste NO2,
AFAIK RFNA is useful for most nitrations, and can be diluted to azeotropic if required.
Members more experienced than myself would perform nitric acid distillations at reduced pressure, hence reduced temperatures,
as nitric acid is decomposed fairly quickly by higher temperatures.
(excess NO2 does not seem to be a problem - it just looks evil)
(which is probably why I like it)
[Edited on 9-4-2019 by Sulaiman]
CAUTION : Hobby Chemist, not Professional or even Amateur
|
|
|
Herr Haber
International Hazard
Posts: 1236
Registered: 29-1-2016
Member Is Offline
Mood: No Mood
|
|
Drop the Vigreux (not literally) it's useless.
It only forces you to use higher temperatures and decompose your product.
|
|
|
nimgoldman
Hazard to Others
Posts: 303
Registered: 11-6-2018
Member Is Offline
|
|
Quote: Originally posted by Sulaiman  | First, it would be simpler to initially add only the quantity of water requied to produce (slightly greater than) azeotropic concentration in the
original reaction,
this way the distillation is not actually required, just removal of the sulphate salt (and other impurities) by cooling and filtration,
A simple distillation would of course give a purer product. |
But how do you tell the reaction is complete? Have you done this filtration or is it just a hypothesis? I have not found this kind of work up
elsewhere.
I thought the reaction starts at high temperatures so at least refluxing for several hours would be needed and if so, then it is as easy to perform
distillation.
This is known to cause significant loss of yield. Yes the nitric acid will be fuming and of higher concentration, but the total amount of HNO3 is
lower. Diluting fuming acid itself produces lots of decomposition and fumes so this is why I like to add water beforehand, not after the fact.
I don't need RNFA as I don't do any nitrations. I need 68% specifically for dissolving metals like silver and making aqua regia. Higher concentrations
don't work for due to passivation of the metal and lower concentrations also work worse due to excess water and thus need for prolonged drying. The
azeotropic one is used in most syntheses I like to reproduce, hence the need for 68%.
| Quote: Originally posted by Sulaiman | Members more experienced than myse
lf would perform nitric acid distillations at reduced pressure, hence reduced temperatures,
as nitric acid is decomposed fairly quickly by higher temperatures. |
Yes I can do vacuum distillation at 100 mbar. I have a chemically resistant pump so this is not an issue.
I didn't know high temperature decomposes nitric acid. I always thought just that the light is the issue.
One problem with vacuum distillation is the shifted boiling points. The crude manometer on the pump can't tell the exact pressure needed to estimate
the boiling point using P-T nomograph and even if pressure is known, it is only an estimate. I have to observe the thermometer the whole time for any
changes as this can be the new fraction coming.
| Quote: Originally posted by Herr Haber | Drop the Vigreux (not literally) it's useless.
It only forces you to use higher temperatures and decompose your product. |
But how do you fractionate then??
Should I use packed column with Rashig rings?
Yes there are some special tray-based fractionating columns but these are huge and very expensive. I know people make 68% nitric acid with only
minimum equipment, in low tech amateur settings (e.g. Doug's Lab on YT). So I know I am making a mistake somewhere and not sure where it is...
Maybe the higher temperature required for Vigreux column and the low performance of it actually leads to lower concentration than simple distillation?
|
|
|
nimgoldman
Hazard to Others
Posts: 303
Registered: 11-6-2018
Member Is Offline
|
|
As for the density calculation, yes I checked the temperature and it was 28 °C. For the density of 1.34 g/mL, the table gives 58% for both 25 °C and
30 °C so I am confident about that concentration as it should fall between the two numbers. But I will do a titration anyway.
I am thinking about using a 40 cm packed column (filled with ceramic rings) and vacuum distillation to concentrate the acid.
|
|
|
WGTR
National Hazard
Posts: 971
Registered: 29-9-2013
Location: Online
Member Is Offline
Mood: Outline
|
|
Can't you get close to 68% nitric acid simply by boiling down the diluted acid? I think that once the concentration gets over 20%, that some acid
would be lost through boiling. If set up for simple distillation these could be recovered, and additional concentrated acid could be recovered by
re-boiling the distillate. It seems like a simple, if inefficient way of doing it.
|
|
|
nimgoldman
Hazard to Others
Posts: 303
Registered: 11-6-2018
Member Is Offline
|
|
| Quote: Originally posted by WGTR | | Can't you get close to 68% nitric acid simply by boiling down the diluted acid? I think that once the concentration gets over 20%, that some acid
would be lost through boiling. If set up for simple distillation these could be recovered, and additional concentrated acid could be recovered by
re-boiling the distillate. It seems like a simple, if inefficient way of doing it. |
Yes, good point. I will do it that way - I will still need vacuum fractional distillation setup, but will only distill off the water, there is no need
to distill over the acid.
In the synthesis, though, the second distillation is beneficial since the product still contains some salts (alkali metal bisulafe?) - once almost all
the acid is distilled, the residue is milky white and the boiling point goes up considerably since the salts concentrate it.
I want to use the acid for organic syntheses as well so I don't want any salts in it, hence the need for two distillations in the original synthesis.
|
|
|
nimgoldman
Hazard to Others
Posts: 303
Registered: 11-6-2018
Member Is Offline
|
|
Okay so I repeated the fractional distillation using vacuum and the separation was still poor.
The liquid started boiling at 66 °C (100 mbar vacuum) and the head temperature was 60 °C meaning there is probably a good separation.
The temperature if the liquid rose to about 72 °C and head temperature to 70-71 °C showing there is no longer a good separation.
I assumed the mixture is already azeotropic and stopped the distillation.
The amount of distillate was about 150 mL which is about the amount of water expected.
Unfortunately the concentration of acid was only 59%. So the distillation removed 10% of the volume, increasing the concentration by about 1% only.
It seems nitric acid cannot be efficiently concentrated with simple or fractional distillation unless very long column is used or
some agent added (like sulfuric acid) to trap the water.
Any ideas?
I think I will lower the amount of water in the initial synthesis to get near-azeotropic acid from start, even at the cost of lower yield, and then
sacrifice 1/10th of the volume of the liquid to get to 68%.
The image shows the setup I used.

|
|
|
UC235
National Hazard
Posts: 565
Registered: 28-12-2014
Member Is Offline
Mood: No Mood
|
|
What is it that you're using to measure density? Density is very temperature sensitive. I would consider a titration to confirm your suspicions.
|
|
|
nimgoldman
Hazard to Others
Posts: 303
Registered: 11-6-2018
Member Is Offline
|
|
| Quote: Originally posted by UC235 | | What is it that you're using to measure density? Density is very temperature sensitive. I would consider a titration to confirm your suspicions.
|
I used two methods, density calculation and titration. Both methods gave me 58% (by titration it was 57.97%) so I am confident about the result.
I used titration to measure acid concentrated by vacuum fractional distillation it was now 59.5%.
For titration, I use 10 g sample which slighly diluted with ddH2O and titrated with 20% NaOH from 50 mL burette (0.1 mL precision) and use few drops
of nitrazine solution as neutralization indicator. The solution is magnetically stirred and the neutralization point is found by sharp color change
which occurs within one drop of addition. I think it is precise to 1%
For density measurement, I use 250 mL volumetric flask (class A) and 0.01 g scale (calibrated).
I will research if small amount of sulfuric acid would help with concentration (I need 68% though, I have no use for fuming nitric acid) and I will
probably try fractional distillation with 80 cm packed column. Hopefully this will finally help. Unfortunately, my packed column tends to overfill so
maybe I will start with 40 cm one and see how it performs.
[Edited on 11-4-2019 by nimgoldman]
[Edited on 11-4-2019 by nimgoldman]
|
|
|
nimgoldman
Hazard to Others
Posts: 303
Registered: 11-6-2018
Member Is Offline
|
|
I just measured the HNO3 concentration in the receiving flask and it was 29%.
Now I realized concentrating the dilute nitric acid all the way to azeotropic may not be achievable so easily, since the at 58%, it boils at 115-118
°C and the column must be able to separate the 2-3 degrees centigrade difference to 21 degrees centigrade difference - that would require a very long
column and multiple distillations.
I think the only viable way would be to add small amount of sulfuric acid to the mixture, as a dehydrating agent, and use simple distillation.
The question is how much sulfuric acid to use? 50% sulfuric acid has a boiling point of 120 °C which is just about the b.p. of azeotropic nitric acid
(121 °C). However, as the water will boill off, the b.p. of the mixture will quickly rise and the azeotropic nitric acid will be the lowest boiling
component. Clearly an excess of sulfuric acid will be needed, as well as re-distilling the mixture and using using fractionation.
I am a bit worried about sulfuric acid contamination though.
[Edited on 11-4-2019 by nimgoldman]
|
|
|
WGTR
National Hazard
Posts: 971
Registered: 29-9-2013
Location: Online
Member Is Offline
Mood: Outline
|
|
There is a book in the Sciencemadness library entitled the “Absorption of Nitrous Gases”. Page 279 begins a chapter on the concentration of
dilute nitric acid by various means, and provides some data that may be useful to you. I have this book, so if there are some pages that are
difficult to read just let me know and I can scan them.
|
|
|
WGTR
National Hazard
Posts: 971
Registered: 29-9-2013
Location: Online
Member Is Offline
Mood: Outline
|
|
From the book, boiling down an approximately 35% solution of nitric acid down to half its volume should give you a 65% concentration in the residue of
the boiling flask. If you did a simple distillation on the diluted acid, then the first 2/3 or so could be your diluted fraction. The next fraction
should be darn near 65-68%, in my understanding.
|
|
|
nimgoldman
Hazard to Others
Posts: 303
Registered: 11-6-2018
Member Is Offline
|
|
I will try that.
| Quote: Originally posted by WGTR | | There is a book in the Sciencemadness library entitled the “Absorption of Nitrous Gases”. Page 279 begins a chapter on the concentration of
dilute nitric acid by various means, and provides some data that may be useful to you. I have this book, so if there are some pages that are
difficult to read just let me know and I can scan them. |
Thanks. This is exactly what I looked for.
I will probably try vacuum distillation to get pure concentrated one and then concentrate the leftovers by adding sulfuric acid and distill them over.
[Edited on 12-4-2019 by nimgoldman]
|
|
|
nimgoldman
Hazard to Others
Posts: 303
Registered: 11-6-2018
Member Is Offline
|
|
Okay I've read the part about concentrating nitric acid from "Absorption Of Nitrous Gases" pp. 279 - 305.
I decided to concentrate my 58% nitric acid to >68% by adding sulfric acid and distill under vacuum. If the resulting nitric acid will be >68%,
I will dilute it afterwards.
There is one interesting graph in the book I don't completely understand:

It seems that adding H2SO4 to my 57% HNO3 (so that final concentration is 10% H2SO4) wil yield
>85% HNO3, right?
I don't understand how long the distillation should run and what is the stopping condition.
There is one graph showing boiling point curves for various mixtures of nitric acid and sulfuric acid, which might give a hint about the stopping
temperature:

From it, this seems that adding H2SO4 to 10% w/w concentration in my 57% nitric acid will raise boiling point to about 120 °C
then slightly going up and then decrease to 110 °C as the nitric acid boils off from the mixture.
But this is problematic, since the dilute sulfuric acid has a lower b.p. than 120 °C and would distill over.
I really need to avoid contaminating nitric acid with sulfuric... I use nitric acid for things like dissolving silver and I really need pure nitrates,
not sulfates.
One solution would be to add excess sulfuric acid (say 60% w.r.t. contained water in the solution), then its b.p. will always be well above the b.p.
of nitric acid at any concentration. This will however produce fuming nitric acid, which I found difficult to dilute without too much decomposition
and NO2 production... it would probably have to be redistilled under vacuum.
I think any distillation carried out should be fractional, but this detail has not been mentioned in the book.
|
|
|
morganbw
National Hazard
Posts: 561
Registered: 23-11-2014
Member Is Offline
Mood: No Mood
|
|
You make me smile. Thank you
|
|
|
WGTR
National Hazard
Posts: 971
Registered: 29-9-2013
Location: Online
Member Is Offline
Mood: Outline
|
|
| Quote: Originally posted by nimgoldman |
But this is problematic, since the dilute sulfuric acid has a lower b.p. than 120 °C and would distill over.
I really need to avoid contaminating nitric acid with sulfuric... I use nitric acid for things like dissolving silver and I really need pure nitrates,
not sulfates. |
The amount of sulfuric acid coming over in a distillation should be almost nothing at 120°C. It's some water that's coming over, with the nitric
acid.
I'm still not convinced that you need sulfuric acid at all anyway. It's Friday. Let me see if I can run a small experiment today. If I can, I'll
post the results.
|
|
|
S.C. Wack
bibliomaster
Posts: 2419
Registered: 7-5-2004
Location: Cornworld, Central USA
Member Is Offline
Mood: Enhanced
|
|
Perhaps you should try to do everything the opposite you think makes sense. Retort still, conc. sulfuric acid, just enough heat for distillate, taking
fractions when heat is increased.
|
|
|
nimgoldman
Hazard to Others
Posts: 303
Registered: 11-6-2018
Member Is Offline
|
|
| Quote: Originally posted by S.C. Wack | | Perhaps you should try to do everything the opposite you think makes sense. Retort still, conc. sulfuric acid, just enough heat for distillate, taking
fractions when heat is increased. |
That's a good idea. I like to use vacuum however, as this produces nice clearless nitric acid (without vacuum, I usually have lots of nitrogen dioxide
produced).
I will probably attach a small fractionating column (just to be on the safe side) do vacuum distillation and collect fractions.
I just realized nitric acid is actually produced from sulfuric acid solution (in the most common synthesis) and there is no noticeable contamination.
|
|
|
nimgoldman
Hazard to Others
Posts: 303
Registered: 11-6-2018
Member Is Offline
|
|
To concentration does not seem to work according to paper. They suggested as little as 10% sulfuric acid will allow producing fuming nitric acid from
fairly dilute one, or I understood the graphs wrong.
Anyway, I took 300 mL of crude 47% nitric acid, added 30 mL of 98% sulfuric acid to it and distilled under vacuum (100 mbar) with a short Vigreux
column. The temperature at still head slowly raised from 61 °C to 68 °C until the temperature slightly dropped and sulfuric acid vapours appeared in
the boiling flask at which point I stopped the distillation.
There was about 270 mL of distillate with a density of 1.3 g/mL corresponding to 47% concentration.
So the addition 10% of the volume in sulfuric acid didn't help in any way. Should I use more sulfuric acid?
Now I am thinking about producing fuming nitric acid by adding excess of sulfuric, then concentrating the near-concentrated nitric acid I have with
the fuming one.
I will probably perform the first distillation (with sulfuric acid) without vacuum to monitor the actual temperature so I can compare it to tables,
then redistill it under vacuum to remove nitric dioxide contamination.
[Edited on 15-5-2019 by nimgoldman]
|
|
|
Keras
National Hazard
Posts: 774
Registered: 20-8-2018
Location: (48, 2)
Member Is Offline
|
|
Why don’t you try producing fuming nitric acid directly from sulphuric acid and potassium nitrate, then you dilute it?
|
|
|
Keras
National Hazard
Posts: 774
Registered: 20-8-2018
Location: (48, 2)
Member Is Offline
|
|
| Quote: Originally posted by nimgoldman | To concentration does not seem to work according to paper. They suggested as little as 10% sulfuric acid will allow producing fuming nitric acid from
fairly dilute one, or I understood the graphs wrong.
Anyway, I took 300 mL of crude 47% nitric acid, added 30 mL of 98% sulfuric acid to it and distilled under vacuum (100 mbar) with a short Vigreux
column. The temperature at still head slowly raised from 61 °C to 68 °C until the temperature slightly dropped and sulfuric acid vapours appeared in
the boiling flask at which point I stopped the distillation.
There was about 270 mL of distillate with a density of 1.3 g/mL corresponding to 47% concentration.
So the addition 10% of the volume in sulfuric acid didn't help in any way. Should I use more sulfuric acid?
[Edited on 15-5-2019 by nimgoldman] |
The curves you posted are pretty clear: here you started with a 50% nitric acid combined with 10% sulphuric acid solution. If you look at the graph,
the rightmost curve which represents 10% sulphuric acid, you will see that you’re not expected to get a much higher concentration of nitric acid in
the fumes (you get even LESS). If you double the sulphuric acid concentration (20%) however, from a 50% nitric acid solution you should be able to
boil off a 80% solution, at a boiling point of around 120 °C, as the second graph tells you. And 30% sulphuric acid should give you almost pure
nitric acid (look up the 50% nitric acid vertical line).
At least that’s the way I interpret those curves.
|
|
|
CobaltChloride
Hazard to Others
Posts: 239
Registered: 3-3-2018
Location: Romania
Member Is Offline
|
|
https://www.sciencemadness.org/whisper/viewthread.php?tid=13... This method is my favorite due to its simplicity. I have done it twice and both
times I got acid with a density that corresponded well to 68%. I only used a molar equivalent of ammonium nitrate instead of KNO3 and then added a few
drops of 50% H2O2 to my HNO3 to remove any residual nitrogen oxides that remained dissolved.
|
|
|
nimgoldman
Hazard to Others
Posts: 303
Registered: 11-6-2018
Member Is Offline
|
|
I've ran the experiment with 900 mL of 47% HNO3. It contains about 595 g of water so I added about half of that volume in sulfuric acid, i.e. 300 mL.
The acid produced was only 58%, not over 80% like the curves in graph suggested.
I then realized most syntheses use as much sulfuric acid as there is water, even more and one synthesis of anhydrous nitric acid from conc. nitric
acid requires five times the volume in sulfuric acid as there is nitric.
So I simply keep the 58% HNO3, it is good enough for most applications (mostly for dissolving metals), but next time doing synthesis, I will simply
use much less water as it is easy to dilute too concentrated nitric acid then to concentrate it later, which wastes the sulfuric acid and a base as
well since we have neutralize all that sulfuric acid...
|
|
|









