| Pages:
1
..
21
22
23
24
25
26 |
B(a)P
International Hazard

Posts: 1132
Registered: 29-9-2019
Member Is Offline
Mood: Festive
|
|
I have had good success making NHN using hydrazine free based from hydrazine sulfate.
Can you describe your process?
My approach has been as follows:
- make sure everything is as dry as possible to start and stir stir stir stir stir then stir the whole way, also keep everything cold at all times.
- add NaOH in two portions
- after the first portion of NaOH add 1 ml of cold DH2O, if the reaction does not take off add another 1 ml.
- add ethanol once the reaction is proceeding
- at this point the solid portion will be sticky, add the second portion of NaOH, this makes the sodium sulfate go to sodium bisulfate and pull a
bunch of water out with it.
- wash twice with ethanol and you have fair concentrated hydrazine in ethanol.
|
|
|
Laboratory of Liptakov
International Hazard
Posts: 1359
Registered: 2-9-2014
Location: Technion Haifa
Member Is Offline
Mood: old jew
|
|
Interesting process. Two dry powders? A like fine powder? Drying NaOH can be problem. Any ratios between Hydrazine sulfate and NaOH? Basic weight of
both compounds? For 1- 2 ml DH2O ? I estimate, vacuum Buchner was used? How much ethanol?
Thanks.
Development of primarily - secondary substances CHP (2015) Lithex (2022) Brightelite (2023) Nitrocelite and KC primer (2024)
|
|
|
MineMan
National Hazard
Posts: 998
Registered: 29-3-2015
Member Is Offline
Mood: No Mood
|
|
He’s back!!
|
|
|
B(a)P
International Hazard
Posts: 1132
Registered: 29-9-2019
Member Is Offline
Mood: Festive
|
|
Quote: Originally posted by Laboratory of Liptakov  | Interesting process. Two dry powders? A like fine powder? Drying NaOH can be problem. Any ratios between Hydrazine sulfate and NaOH? Basic weight of
both compounds? For 1- 2 ml DH2O ? I estimate, vacuum Buchner was used? How much ethanol?
Thanks. |
Sorry for the slow reply, I missed your message. I just dug out out my lab notes from last time I did this.
10 g of hydrazine sulfate
17 g EtOH start with 10 g then use 7 g for the second rinse
6.15 g NaOH added in two equal portions as described above
1.5 mLs dH2O
I did not use a funnel or filtration I simply decanted the ethanol/hydrazine liquid from the sodium bisulfate once it had settled.
Edit - Yes both solids as fine dry powders stored in a desiccator bag prior to use.
[Edited on 16-11-2020 by B(a)P]
|
|
|
Laboratory of Liptakov
International Hazard
Posts: 1359
Registered: 2-9-2014
Location: Technion Haifa
Member Is Offline
Mood: old jew
|
|
Thanks BP....interest method...
Development of primarily - secondary substances CHP (2015) Lithex (2022) Brightelite (2023) Nitrocelite and KC primer (2024)
|
|
|
dangerous amateur
Hazard to Others
Posts: 148
Registered: 8-7-2011
Member Is Offline
Mood: No Mood
|
|
Sorry for the late answer 
Sadly I did not find my old notes, but I used Barium hydroxide back then. But I concluded that it's not working
I will try it again this year.
| Quote: |
10 g of hydrazine sulfate 17 g EtOH start with 10 g then use 7 g for the second rinse 6.15 g NaOH added in two equal portions as described above 1.5
mLs dH2O |
Could you somehow judge the yield somehow? How do you know it was
?
[Edited on 6-2-2021 by dangerous amateur]
|
|
|
Hens2
Harmless
Posts: 1
Registered: 29-7-2019
Member Is Offline
|
|
You can weigh out the sodium sulfate thats left behind and do some maths. calculate the theoretical amount of sodium sulfate which should be
synthesised in the procedure. than check it with the one that was left behind. dont forget to add the small amount which is desolved in etoh
(solubility of na2so4 is about 0.44g/100g of etoh (100%)). with that you should be able to calculate the mass percentage of the hydrazine in your etoh
solution.. correct me if i am wrong.
|
|
|
night429
Harmless
Posts: 48
Registered: 12-11-2019
Member Is Offline
Mood: 
|
|
I've seen it mentioned once in this thread, but I made (what I believe to be) tetraamminecopper (ii) picrate. I made this by adding a solution of
excess tetraamminecopper (ii) sulfate to a solution of warm ammonium picrate with a bit of ammonia to ensure the complex didn't fall apart.
Immediately, a flaky gold-colored precipitate formed. Upon filtering, the filtrate remained the typical color of the tetraamminecopper (ii) ion,
indicating that the complex hadn't fallen apart. Drying caused the precipitate to "mat", and I chopped it up while damp with a metal spatula.

The properties of the product are typical with both a copper salt and a picrate. Upon exposure to flame, a green color is observed, indicating copper
is present. The product then melts, followed by vigorous and rapid burning; a noise and flame are both present. To prove the compound I made wasn't
just copper (ii) picrate, I made the latter by adding copper (ii) chloride to a concentrated sodium picrate solution. A green precipitate formed,
which was filtered. The properties of this precipitate, not only color, are different than the compound I made. Burning is less vigorous, but it burns
more readily and doesn't melt beforehand.
Comparison of the supposed tetraamminecupric picrate (left) and cupric picrate (left):

So, the only conclusion I could come up with was that the product I have is tetraamminecopper(ii) picrate, considering its properties are quite
different than both ammonium picrate and copper (ii) picrate, and I don't think that a product could've been made that wasn't this one, given the
conditions I performed the reaction in.
"What God would damn a heart?"
|
|
|
Raid
Hazard to Everyone
Posts: 201
Registered: 14-11-2022
Location: N/A
Member Is Offline
Mood: School
|
|
i did the same thing as night429 but instead of the sulfate I did a nitrate and it makes a nice dark forest green complex. its somewhat energetic, I'm
going to do some more large scale tests with it later.
|
|
|
Raid
Hazard to Everyone
Posts: 201
Registered: 14-11-2022
Location: N/A
Member Is Offline
Mood: School
|
|
I found this in a patent for NiHN and CoHN and it says that adding KCLO3 or LFCN (Lead Ferrocyanide) reduces the friction sensitivity, I'll post a
screenshot of the chart here if you want to see it. I'm wondering if this is even worth doing since they don't show the effects on DDT velocity. They
did say that the NiHN is an almost oxygen balanced compound and I'm thinking that maybe the KCLO3 may help the DDT a bit, but since the KCLO3 is not
Incorporated in the molecule i'm not so sure.
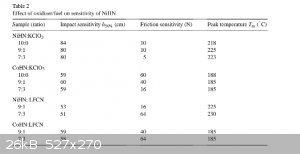
|
|
|
papaya
National Hazard
Posts: 615
Registered: 4-4-2013
Member Is Offline
Mood: reactive
|
|
I've created a whole new thread since your question raised some memories... btw can you provide a link to that patent, since it sounds like parallel
to my own experiments? https://www.sciencemadness.org/whisper/viewthread.php?tid=15...
[Edited on 31-3-2023 by papaya]
|
|
|
Raid
Hazard to Everyone
Posts: 201
Registered: 14-11-2022
Location: N/A
Member Is Offline
Mood: School
|
|
I have uploaded the patent in the "NHN based improved primary composition" that you have just made.
|
|
|
papaya
National Hazard
Posts: 615
Registered: 4-4-2013
Member Is Offline
Mood: reactive
|
|
| Quote: Originally posted by Raid | | I have uploaded the patent in the "NHN based improved primary composition" that you have just made. |
Thanks
a lot!
|
|
|
specialactivitieSK
Hazard to Self
Posts: 94
Registered: 21-10-2014
Member Is Offline
Mood: No Mood
|
|
Can 500g TACN ignite standard granulated ANFO?
[Edited on 23-4-2023 by specialactivitieSK]
|
|
|
MineMan
National Hazard
Posts: 998
Registered: 29-3-2015
Member Is Offline
Mood: No Mood
|
|
Can this get removed? It puts this place in a bad name and is low effort. Has nothing to do with exotic primaries which are used sub gram
|
|
|
Laboratory of Liptakov
International Hazard
Posts: 1359
Registered: 2-9-2014
Location: Technion Haifa
Member Is Offline
Mood: old jew
|
|
I am also for remove 500g TACN question.
Development of primarily - secondary substances CHP (2015) Lithex (2022) Brightelite (2023) Nitrocelite and KC primer (2024)
|
|
|
dettoo456
Hazard to Others
Posts: 218
Registered: 12-9-2021
Member Is Offline
|
|
I may or may not have made some sodium 3-nitro-1,2,4-triazolate recently and now I can’t figure out what to do with it. It is reasonably powerful on
its own (unconfined heating from under Al foil causes melting and a flash/poof similar to organic peroxide ignition). The Ag salt is like more
powerful tetrazene - same smokey, poofy decomp but can actually deflagrate or detonate in the right confinement. I want to make the Cu (i) and (ii)
salts when I get a chance and they should act as a sort of insensitive booster EM.
Anyways though, I wanted to know if making the triazole variant of BNCP would likely bear any fruit? It’s already extremely stable, so would
replacing the 1 N with C just make it kind of overly insensitive and therefore useless?
|
|
|
Microtek
National Hazard
Posts: 845
Registered: 23-9-2002
Member Is Offline
Mood: No Mood
|
|
I would request that you examine solubilities of the salt as well as the free acid (I know that doesn't sound very exciting, but many contemporary
papers don't bother to do that, and it can really help with preparation and identification if you know how soluble your product is). Then I would be
interested in salts of nitrogen containing bases (ammonia, hydrazine, hydroxylamine, etc.). How did you synthesize the material?
|
|
|
dettoo456
Hazard to Others
Posts: 218
Registered: 12-9-2021
Member Is Offline
|
|
Since I had some old, yellowing AGB lying around, I refluxed it in >90% CH2O2 for about 3-4hrs until the acid smell stopped and the mixture was a
yellow, transparent liquid. Then I evaporated it down under gentle heat (this took a long time and even then, the final off-white goop formed a
hygroscopic gel - I assume this material is 3-amino-1,2,4-triazole with impurities of formylaminoguanidine.
With the assumption being by that the product is mostly Amitrole, I followed the PacSci Oxidative nitrosation in US9598380B2 (prep for 5-NaNTz) but
modified by mole scale to 3-Amino-1,2,4-triazole. The reaction was about the same as for 5-ATz but it generated what seemed like larger amounts of
NOx. The product (assumed to be Sodium 3-nitro-1,2,4-triazolate) precipitated in the crude rxn mix even in stirring and above 60C. Rxn miz was then
allowed to cool, product filtered, dried, then extracted with Acetone to get rid of Na2SO4 and bisulfate.
The final product (Na-3-NTiz), crystallizing out of acetone, presents as bright yellow, opaque ‘petal-like’ shapes. It does seem to be modestly
hygroscopic on standing in an open vessel at rt.
I’m busy right now but I’ll try to give more info and pictures later on.
[Edited on 1-6-2023 by dettoo456]
|
|
|
Microtek
National Hazard
Posts: 845
Registered: 23-9-2002
Member Is Offline
Mood: No Mood
|
|
I think you are supposed to react the product of the acetylization (from heating the guanidine derivative with carboxylic acid) with aqueous base.
Sodium- or potassium carbonate seem popular from the papers I have read.
Attachment: kurzer1963(1).pdf (1.6MB)
This file has been downloaded 202 times
|
|
|
dettoo456
Hazard to Others
Posts: 218
Registered: 12-9-2021
Member Is Offline
|
|
I know that Amitrole is produced industrially via formylation and cyclization of AGB, but there are very few papers on the prep of 3-nitrotriazole.
One paper (https://onlinelibrary.wiley.com/doi/10.1002/047084289X.rn020...) lays out a synth starting from amitrole but i had issues in the past trying to
replicate it, so I used the modified 5-ATz oxidative nitrosation route.
Also, I edited the last post I made - the Sodium salt IS hygroscopic, or at least forms a type of hydrate from crystallizing in acetone.
I also don’t know how to post pics or videos in SM, if you could let me know how I’d be very grateful. Thanks
[Edited on 1-6-2023 by dettoo456]
[Edited on 1-6-2023 by dettoo456]
|
|
|
articneptune
Harmless
Posts: 2
Registered: 5-9-2023
Member Is Offline
|
|
Hello all,
I'm currently working on synthesis of hydroxylammonium nitrate HAN. I've got it setup as per the attachment but using 70% HNO3 instead of 65%.
I've had one attempt at it so far, but I couldn't keep the mixture cool enough and it started reacting quite violently. I'm going to give it another
shot soon but with salt in the ice bath. I'm hoping this will keep it cool enough. Once I've got HAN I'm planning on adding some methanol and AN to
make something like HAN269MEO (rocket monopropellant). If there's any left over I might just put it in the rotary evap and try get some HAN salt.
A question I have is how would you add the methanol and AN? Just measure it out and mix it in?
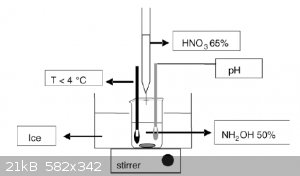
|
|
|
Microtek
National Hazard
Posts: 845
Registered: 23-9-2002
Member Is Offline
Mood: No Mood
|
|
Yes, I would simply measure out AN and MeOH. Isn't this setup unnecessarily complicated? I would just add a slight excess of HNO3 and then neutralize
the excess with a little ammonia. If you keep the mix well stirred and either add the acid very slowly (dropwise) or dilute it a little more, I doubt
you will have any trouble with excessive exothermicity.
|
|
|
articneptune
Harmless
Posts: 2
Registered: 5-9-2023
Member Is Offline
|
|
Thanks for the reply
It may be yes but it's a procedure used in literature that I could reference if I ever need to. I've since managed to make a batch of HAN. I'm going
to be mixing it with various things and putting it in a strand burner this week. Left over HAN is going to be concentrated to it's salt form and then
ignited. I'll update if anything particularly interesting occurs.
|
|
|
dettoo456
Hazard to Others
Posts: 218
Registered: 12-9-2021
Member Is Offline
|
|
Anyone had experience with Silver 5-ATz complexes such as [(2Ag)5-ATz] NO3 or ClO4? The perchlorate is mentioned in engager’s tetrazole write up and
a patent but there doesn’t seem to be much discussion on it. I tried to make the nitrate the other day according to klapotke’s procedure (https://pubmed.ncbi.nlm.nih.gov/19105192/) but it didn’t seem to yield anything that great. To start, 5-ATZ doesn’t dissolve that great in
conc (anything higher than azeo %) Nitric Acid, and if you add too much water, the complexation time with AgNO3 can be ruined by just dumping out the
Silver salt of 5-ATz. Too high conc of acid will form the 5-ATz nitrate salt instead which has low solubility as well and hinders complexation as
well.
My product crashed out of the sol immediately on addition of AgNO3 to the 5-ATz/HNO3 mix as an off-white precipitate that started to yellow to a
pastel yellow on air drying on a filter.
The product is supposed to detonate on flame contact and indirectly at 298C. For me, it does crackle faintly but I can’t seem to get it to dry. I
would wager it’s like AgNTz in that it is a pain to detonate when wet but really goes when dry, but I can’t get this complex to dry to see
anything amazing.
This salt could be a great alternative to LA, nitrotetrazoles, and such since it’s so seemingly easy to prepare, but I can’t get make it to match
klapotke’s results. -My HNO3 and AgNO3 are pure and my 5-ATz is decently pure so idk.
|
|
|
| Pages:
1
..
21
22
23
24
25
26 |








